Решение Коллегии Евразийской экономической комиссии от 14.01.2020 N 1
КОЛЛЕГИЯ ЕВРАЗИЙСКОЙ ЭКОНОМИЧЕСКОЙ КОМИССИИ
РЕШЕНИЕ
от 14 января 2020 г. N 1
ОБ УТВЕРЖДЕНИИ РУКОВОДСТВА
ПО УСТАНОВЛЕНИЮ ДОПУСТИМЫХ ПРЕДЕЛОВ ВОЗДЕЙСТВИЯ НА ЗДОРОВЬЕ
В ЦЕЛЯХ ИДЕНТИФИКАЦИИ РИСКОВ ПРИ ПРОИЗВОДСТВЕ ЛЕКАРСТВЕННЫХ
СРЕДСТВ НА ОБЩИХ ПРОИЗВОДСТВЕННЫХ (ТЕХНОЛОГИЧЕСКИХ) ЛИНИЯХ
В соответствии со статьями 30 и 56 Договора о Евразийском экономическом союзе от 29 мая 2014 года, пунктом 14 Протокола о применении санитарных, ветеринарно-санитарных и карантинных фитосанитарных мер (приложение N 12 к Договору о Евразийском экономическом союзе от 29 мая 2014 года), статьей 4, пунктом 7 статьи 7, пунктом 1 статьи 9 Соглашения о единых принципах и правилах обращения лекарственных средств в рамках Евразийского экономического союза от 23 декабря 2014 года и пунктом 5 перечня актов Евразийской экономической комиссии по вопросам регулирования общих рынков лекарственных средств и медицинских изделий в рамках Евразийского экономического союза на 2017 - 2019 годы (приложение к распоряжению Совета Евразийской экономической комиссии от 17 мая 2017 г. N 15) Коллегия Евразийской экономической комиссии решила:
1. Утвердить прилагаемое Руководство по установлению допустимых пределов воздействия на здоровье в целях идентификации рисков при производстве лекарственных средств на общих производственных (технологических) линиях.
2. Настоящее Решение вступает в силу по истечении 6 месяцев с даты его официального опубликования.
Председатель Коллегии
Евразийской экономической комиссии
Т.САРКИСЯН
Утверждено
Решением Коллегии
Евразийской экономической комиссии
от 14 января 2020 г. N 1
РУКОВОДСТВО
ПО УСТАНОВЛЕНИЮ ДОПУСТИМЫХ ПРЕДЕЛОВ ВОЗДЕЙСТВИЯ НА ЗДОРОВЬЕ
В ЦЕЛЯХ ИДЕНТИФИКАЦИИ РИСКОВ ПРИ ПРОИЗВОДСТВЕ ЛЕКАРСТВЕННЫХ
СРЕДСТВ НА ОБЩИХ ПРОИЗВОДСТВЕННЫХ (ТЕХНОЛОГИЧЕСКИХ) ЛИНИЯХ
I. Общие положения
1. Настоящее Руководство применяется для анализа и оценки рисков при производстве разных, лекарственных средств, в том числе высокоактивных на общих производственных (технологических) линиях с целью установления допустимых пределов воздействия на здоровье.
2. При производстве разных лекарственных средств на общих производственных (технологических) линиях существует потенциальная опасность их перекрестной контаминации.
3. В случае если одно лекарственное средство в процессе совместного производства контаминирует другое лекарственное средство, его необходимо рассматривать с точки зрения создаваемого им риска для человека или животного, независимо от его положительного воздействия при применении по прямому предназначению. Присутствие таких контаминантов необходимо контролировать в соответствии с риском, который они представляют. Риск определяется содержанием контаминирующего вещества, которое можно признать безопасным для всех популяций. В целях идентификации возникающих рисков следует использовать допустимые пределы воздействия на здоровье, являющиеся производными безопасного порогового значения для содержания контаминирующего вещества. Применяемые производителем инструменты подхода к идентификации рисков должны быть основаны на использовании научной оценки фармакологических и токсикологических данных для критериев оценки: допустимой ежедневной экспозиции (PDE), пороге токсикологической угрозы (TTC), приемлемой ежедневной экспозиции (ADE), предельной экспозиции на рабочем месте (OEL), что позволяет проводить анализ рисков и контроль экспозиции продуктов перекрестной контаминации для человека.
4. Инструменты подхода к идентификации рисков при производстве лекарственных средств на общих производственных (технологических) линиях, представленного в настоящем Руководстве, разработаны в соответствии с Правилами надлежащей производственной практики Евразийского экономического союза, утвержденных Решением Совета Евразийской экономической комиссии от 3 ноября 2016 г. N 77 (далее - Правила производственной практики).
5. При достаточном обосновании для установления безопасных пороговых значений, указанных в пункте 4 настоящего Руководства, допустимо отклонение от основного подхода, описанного в разделе IV настоящего Руководства.
6. В процессе производства лекарственных средств к случайной перекрестной контаминации приводят неконтролируемый выброс пыли, газов, паров, аэрозолей, генетического материала или организмов:
из активных фармацевтических субстанций, других исходных материалов, а также других видов продукции, обрабатываемых одновременно;
из остаточных загрязнений на оборудовании;
от одежды операторов.
Вследствие возможного риска производство определенных классов лекарственных средств (включая определенные антибиотики, определенные гормоны, определенные цитотоксические вещества и определенные высокоактивные лекарственные средства) требуется проводить в специально отведенных или выделенных помещениях и на специальном оборудовании.
7. В процессе производства лекарственных средств для снижения риска контаминации необходимо использовать очистку и валидацию процесса очистки на основе применения таких пороговых значений контаминирующих факторов в исследованиях валидации процесса очистки, которые допустимы для переноса в новый производственный процесс. Для установления таких норм используют подходы, которые не учитывают доступные фармакологические и токсикологические данные. Следовательно, требуется индивидуальный подход к идентификации рисков и поддержанию мер по снижению рисков для всех классов фармацевтических субстанций.
8. Целью настоящего Руководства является представление подхода к анализу и оценке фармакологических и токсикологических данных отдельных активных фармацевтических субстанций и обеспечение возможности установления пороговых значений (норм), для того чтобы обеспечить соответствие производственного процесса требованиям Правил производственной практики. Пороговые значения допускается использовать в качестве:
инструмента идентификации рисков;
обоснования предельных значений переносимых остатков действующего вещества (контаминанта) в последующий производственный процесс, при валидации процесса очистки.
В соответствующих случаях допускается использовать общие принципы, изложенные в настоящем Руководстве, в целях получения порогового значения для идентификации таких рисков при производстве активных фармацевтических субстанций.
II. Область применения
9. Областью применения настоящего Руководства является обеспечение безопасности пациентов и целевых продуктивных животных, подвергающихся воздействию остаточных количеств действующих веществ (контаминантов) при приеме лекарственных препаратов, а также потребителей, потенциально подвергаемых влиянию остаточных количеств действующих веществ (контаминантов), присутствующих в продуктах питания животного происхождения в результате лечения целевых продуктивных животных ветеринарными лекарственными препаратами, в которых содержатся остаточные количества действующих веществ (контаминантов). Настоящее Руководство устанавливает подход к получению научно обоснованного порогового значения для отдельных действующих веществ, подлежащего применению в целях идентификации рисков. В целях применения единого подхода для любого производственного процесса в рамках фармацевтической отрасли в настоящем Руководстве описывается способ представления данных, которые лежат в основе установления порогового значения.
III. Определения
10. Для целей настоящего Руководства используются понятия, которые означают следующее:
"действующее вещество" - активная фармацевтическая субстанция, входящая в состав лекарственного средства;
"доза, не оказывающая видимого нежелательного эффекта" (No-Observed Adverse Effect Level, NOAEL) - наибольшая концентрация или количество действующего вещества, не оказавшее в эксперименте нежелательное влияние на морфологию, функционирование, рост, развитие или продолжительность жизни организма-мишени, которое можно было бы отличить от аналогичных параметров у контрольных организмов-мишеней, относящихся к тому же биологическому виду и линии при заданных условиях экспозиции (выраженных в виде массы активной фармацевтической субстанции в миллиграммах на 1 килограмм массы тела организма-мишени);
"допустимая ежедневная экспозиция" (Permitted Daily Exposure, PDE) - максимальная допустимая доза конкретного действующего вещества, не вызывающая негативных последствий при ежедневном применении в течение всей жизни человека;
"наименьшая доза, оказывающая явный нежелательный эффект" (Lowest Observed Adverse Effect Level, LOAEL) - наименьшая концентрация или количество действующего вещества, оказавшее в эксперименте нежелательное влияние на морфологию, функционирование, рост, развитие или продолжительность жизни организма-мишени, которое можно было бы отличить от аналогичных параметров у контрольных организмов-мишеней, относящихся к тому же биологическому виду и линии при заданных условиях экспозиции;
"порог токсикологической угрозы" (Threshold of Toxicological Concern, TTC) - генотоксический уровень экспозиции примеси, который приводит к теоретическому канцерогенному риску, равному 1 дополнительному случаю онкологического заболевания на 100 000 пациентов при пожизненной экспозиции.
IV. Установление пределов воздействия на здоровье человека
1. Расчет допустимой ежедневной экспозиции (PDE)
11. Процедура, описанная в настоящем Руководстве, для установления пределов допустимого воздействия на здоровье человека по остаточным количествам действующего вещества (контаминанта) основывается на методе расчета допустимой ежедневной экспозиции (PDE). PDE представляет собой специфичную для вещества дозу, которая не вызовет нежелательный эффект (если индивид на протяжении всей жизни подвергается ежедневной экспозиции в дозе, равной ей или ниже).
Расчет PDE предусматривает:
выявление рисков путем анализа всех значимых данных;
определение критических эффектов;
установление NOAEL, в отношении явлений, признанных критическими эффектами;
использование ряда поправочных коэффициентов для учета влияния различных видов неопределенности.
PDE рассчитывается по формуле:
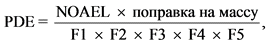
где:
NOAEL - доза, не оказывающая видимого нежелательного эффекта;
F1 - коэффициент, учитывающий поправку на экстраполяцию между видами животных;
F2 - коэффициент, учитывающий межиндивидуальную вариабельность;
F3 - коэффициент, учитывающий исследования токсичности с многократным дозированием короткой продолжительности (то есть менее 4 недель);
F4 - коэффициент, который применяется в случаях опасных видов токсичности действующего вещества (контаминанта) (например, негенотоксической канцерогенности, нейротоксичности или тератогенности);
F5 - переменный коэффициент, который применяется в случае если не установлена доза, не оказывающая эффекта.
12. Для ветеринарных лекарственных средств используется PDE-подход для установления допустимых пределов воздействия на здоровье животного на основе установления предельных значений контаминирующих факторов у разных целевых видов животных. Значения пределов воздействия на здоровье, установленных для животных, не используются для установления пределов воздействия экспозиций действующих веществ (контаминантов) на здоровье человека.
13. Необходимым подходом к установлению пределов воздействия на здоровье человека является расчет PDE на основании данных об экспозиции действующего вещества (контаминанта) у человека. В этом случае допустимая степень контаминации рассчитывается на основании PDE для человека, даже если контаминируемое лекарственное средство является ветеринарным лекарственным средством. Такой подход аналогичен подходу, при котором PDE используется для расчета предельного содержания остаточных растворителей в лекарственном средстве, применяемому при производстве ветеринарных лекарственных средств.
14. При определении предельных значений действующего вещества (контаминанта) необходимо учитывать вводимую дозу лекарственного препарата, которая будет зависеть от массы тела используемого вида животного. Для этого при расчете PDE необходимо использовать значение дозы выраженное в виде мг/кг массы тела животного (то есть использовать коэффициент поправки на массу, равный 1), не в виде значения дозы для всего животного в целом. Если в информации о лекарственном средстве, которое выпускается на производственной (технологической) линии после лекарственного средства для которого рассчитывается предел воздействия на здоровье человека, указывается суточная доза для пациента, а не доза в мг/кг массы тела, то для расчета дозы лекарственных средств для медицинского применения переведенной в мг/кг массы тела следует использовать значение массы тела в 50 кг принятое за стандартную массу тела человека. Для лекарственных средств для ветеринарного применения дозы указываются в мг/кг массы тела. Если это невозможно, следует исходить из массы тела, равной 1 кг, поскольку она будет отражать нижнюю границу массы тела животного.
15. Если лекарственное средство может подвергнуться контаминации остаточным количеством действующего вещества и является ветеринарным лекарственным средством, вводимым продуктивным животным, то используемое предельное значение переноса действующего вещества (контаминанта) должно учитывать как безопасность релевантного вида животного, так и безопасность потребителей. В связи с этим необходимо подтвердить с помощью моделирования сценария наихудшей экспозиции действующего вещества (контаминанта), что ни релевантный вид животного, ни потребитель не подвергнутся экспозиции остаточным количеством действующего вещества, превышающим PDE.
16. Допускается использовать подходы, альтернативные определению NOAEL при установлении явного нежелательного эффекта, такие как ориентировочная доза (Benchmark dose).
17. Допускается использовать иные подходы при обосновании установления допустимых пределов воздействия на здоровье при представлении их научного обоснования.
Требования, предъявляемые к токсикологическим
и фармакологическим данным необходимым
для выявления опасностей
18. Выявление опасностей - это качественная оценка индивидуальных свойств действующего вещества (контаминанта), способных вызывать нежелательные эффекты. С целью выявления опасностей необходимо провести анализ всех доступных данных о каждом действующем веществе (контаминанте) у животных и человека. К данным, необходимым для выявления опасностей относятся:
доклинические фармакодинамические данные;
исследования токсичности с многократным дозированием;
исследования канцерогенности;
исследования генотоксичности in vitro и in vivo;
исследования репродуктивной и онтогенетической токсичности;
клинические данные (терапевтические и нежелательные эффекты).
Доступность данных о действующем веществе будет варьироваться в зависимости от стадии разработки лекарственного препарата и показания к его применению. Если данные неполные, то выявленные пробелы потребуют критической оценки с точки зрения влияния отсутствия таких данных на установление допустимых пределов воздействия на здоровье человека.
Идентификация критических эффектов
19. К критическим эффектам относятся наиболее чувствительные к изменению дозы действующего вещества (контаминанта) нежелательные эффекты, выявленные в доклинических токсикологических исследованиях, при отсутствии доказательств (например, по результатам проведения исследований in vitro, при наличии фармакодинамических данных и т.д.), что такие сведения незначимы для человека или целевых животных. К критическим эффектам также относятся любые фармакологические и нежелательные эффекты.
Установление значения NOAEL
20. Для всех выявленных критических эффектов необходимо установить NOAEL.
21. Если критический эффект отмечается в нескольких исследованиях на животных, для расчета значения PDE следует использовать NOAEL, регистрируемую при наименьшей дозе действующего вещества (контаминанта).
22. Если NOAEL неизвестна, допускается использовать наименьшую дозу, оказывающую явный нежелательный эффект (LOAEL).
23. NOAEL, основанная на клинических фармакодинамических эффектах, должна соответствовать высшей испытанной дозе, признанной терапевтически неэффективной.
Использование поправочных коэффициентов
24. PDE рассчитывается путем деления значения NOAEL установленного для критического эффекта на разные поправочные коэффициенты (коэффициенты безопасности, коэффициенты неопределенности, коэффициенты оценки) для учета влияния различных видов неопределенности и для обеспечения экстраполяции значения NOAEL на достоверную и надежную дозу действующего вещества (контаминанта), не вызывающую явного нежелательного эффекта у человека или релевантного вида животного.
25. Поправочные коэффициенты F1 - F5 учитывают следующие источники неопределенности:
F1 - коэффициент (значения которого находятся в диапазоне 2 - 12) поправки на экстраполяцию между видами животных;
F2 - коэффициент (равный 10), учитывающий межиндивидуальную вариабельность;
F3 - коэффициент (равный 10), учитывающий исследования токсичности с многократным дозированием короткой продолжительности (то есть менее 4 недель);
F4 - коэффициент (1 - 10), который применяется в случаях развития тяжелых токсических реакций (например, негенотоксической канцерогенности, нейротоксичности или тератогенности);
F5 - переменный коэффициент, который применяется в случае если не установлена доза, не оказывающая эффекта. Если известна только наименьшая доза, оказывающая явный нежелательный эффект, в зависимости от тяжести токсического действия действующего вещества (контаминанта) используют коэффициент вплоть до 10.
26. Использование дополнительных поправочных коэффициентов учета остаточной неопределенности, не охваченной коэффициентами перечисленными в пункте 25 настоящего Руководства, допустимо, если они обоснованы научными данными и представлен достаточный анализ, обосновывающий их применение (например, при отсутствии данных о репродуктивной и онтогенетической токсичности действующего вещества (контаминанта) (подраздел 4 раздела V настоящего Руководства)).
27. Использование поправочных коэффициентов должно быть обосновано. Допускается руководствоваться указаниями по выбору поправочных коэффициентов F1 и F4, приведенными в научных руководствах по токсикологии, при этом в обосновании стратегии установления допустимой ежедневной экспозиции (PDE) (в соответствии с приложением к настоящему Руководству) следует приводить библиографическую ссылку на такие руководства.
28. При расчете PDE основанном на токсикологических данных полученных у человека допустимо отказаться от использования коэффициента F2 и (при наличии научного обоснования) коэффициента F5. При достаточном научном обосновании могут использоваться иные поправочные коэффициенты, значения которых отличаются от значений, установленных по умолчанию для поправочных коэффициентов, приведенных в пункте 25 настоящего Руководства.
Выбор окончательного значения PDE
29. Если обнаружено несколько критических эффектов, приводящих к расчету нескольких значений PDE, необходимо представить соответствующее обоснование и выбрать наиболее подходящую PDE для использования ее при валидации процесса очистки. По умолчанию используется минимальное из полученных значений PDE.
2. Использование клинических данных
30. Целью установления допустимых пределов воздействия на здоровье является обеспечение безопасности человека, поэтому особое значение имеет надлежащее качество клинических данных. Нежелательные фармакодинамические эффекты у пациентов, вызванные контаминацией действующими веществами, могут представлять опасность, поэтому при определении критического эффекта необходимо учитывать клинические фармакологические данные. Необходимо учитывать, каким образом рассматриваемое действующее вещество было связано с развитием критического нежелательного эффекта в клинических условиях.
31. Если критический эффект, определенный для установления пределов воздействия на здоровье, основывается на фармакологических и (или) токсикологических эффектах, выявленных у человека, а не у животных, использование формулы для расчета PDE может быть некорректным, поэтому для достижения этой цели допустима оценка клинических данных специфичных для действующего вещества (контаминанта).
Экстраполяция рассчитанного значения PDE для одного пути
введения на другие пути введения
32. Несмотря на то, что значение PDE, рассчитываемое для действующего вещества (контаминанта) основывается на исследованиях с целевым клиническим путем введения, путь введения действующего вещества или лекарственного средства, которое производится производственной (технологической) линии после действующего вещества или лекарственного средства для которого рассчитано PDE, может быть иным. Изменение пути введения может изменить биодоступность действующего вещества (контаминанта), поэтому если есть явные различия (например, различия > 40%) в биодоступности, присущей изученному в токсикологических исследованиях пути введения, для экстраполяции токсикологических данных с одного пути введения на другой необходимо использовать поправочные коэффициенты. Поскольку биодоступность действующего вещества (контаминанта) у разных видов животных может отличаться, поправочные коэффициенты для экстраполяции токсикологических данных с одного пути на другой должны основываться на данных, полученных у человека или на данных полученных у релевантного вида животного - в отношении ветеринарных лекарственных средств.
33. Если данные о биодоступности действующего вещества (контаминанта) для других путей введения у человека или релевантного вида животного отсутствуют и ожидается, что смена пути введения (например, с перорального на ингаляционный) может привести к повышенной системной экспозиции действующего вещества (контаминанта), допускается провести экстраполяцию токсикологических данных с использованием консервативного подхода (исходя из 100% биодоступности действующего вещества (контаминанта)). Например, в случае экстраполяции токсикологических данных с перорального пути на ингаляционный (для которого респирабельная абсорбция составляет 100%), PDE, полученную на основании пероральных данных, можно скорректировать, умножая ее на поправочный коэффициент, который рассчитывается по следующей формуле:

Если данные о биодоступности действующего вещества (контаминанта) у человека или релеватного вида животного для других путей отсутствуют и ожидается, что системная экспозиция контаминанта будет ниже при пути, с помощью которого вводится контаминированное действующее вещество (лекарственное средство), при расчете PDE нет необходимости использовать поправочный коэффициент. Экстраполяцию данных, полученных для одного пути введения на другой следует осуществлять в отдельно для каждого случая.
V. Особые случаи
1. Действующие вещества, обладающие
генотоксическим потенциалом
34. Экспозиция генотоксичных действующих веществ, не имеющих различимого порога токсикологической угрозы, несет риск воздействия на здоровье человека. Вместе с тем предварительно заданный уровень допустимого риска для беспороговых генотоксикантов в форме порога токсикологической угрозы (TTC), составляет 1,5 мкг/индивид/сут. Принимая во внимание, что продолжительность экспозиции остаточных действующих веществ будет существенно более ограниченной (например, поскольку на практике степень переноса остаточных действующих веществ будет снижаться от серии к серии), предельные значения, основанные на максимальной экспозиции, равной 1,5 мкг/индивид/сут, в этом случае не будут превышать теоретический канцерогенный риск, равный 1 x 10-6. Следовательно, в отношении беспороговых остаточных действующих веществ допускается применять предельную дозу, равную 1,5 мкг/индивид/сут.
35. Если лекарственное средство, потенциально контаминированное остаточным действующим веществом, является ветеринарным лекарственным средством, следует использовать тот же TTC, но выражать его "на кг массы тела" (то есть, TTC составляет 0,03 мкг/кг массы тела/день). Если контаминированная продукция предназначена для введения продуктивным животным, при определении предела переноса необходимо учитывать требования по обеспечению безопасности животных, так и требования по обеспечению безопасности для потребителей продуктов, получаемых от этих животных. Следует подтвердить, исходя из наихудших сценариев экспозиции, что ни релевантный вид животного, ни потребители продуктов, получаемых от таких животных не будут подвергаться экспозиции остаточных уровней действующего вещества, превышающих TTC.
36. В отношении генотоксичных действующих веществ в случае достаточного количества данных об их канцерогенности следует применять оценки риска для конкретных соединений вместо TTC.
37. В отношении генотоксичных действующих веществ с достаточными доказательствами порогового механизма безопасные значения экспозиции могут быть установлены с использованием подхода основанного на расчете PDE без заметного риска генотоксичности.
2. Действующие вещества с высоким
сенсибилизирующим потенциалом
38. У людей с повышенной восприимчивостью действующему веществу (контаминанту) могут развиться иммуноопосредованные реакции гиперчувствительности на лекарственные препараты вплоть до потенциально летальных анафилактических реакций.
39. В соответствии с пунктом 3.6 главы 3 Правил производственной практики необходимо предусматривать выделенные помещения и оборудование для производства активных фармацевтических субстанций и лекарственных средств, обладающих высоким сенсибилизирующим потенциалом, научные данные о которых свидетельствуют об отсутствии приемлемой продолжительности экспозиции или риска, возникающего при работе с лекарственным средством на производственной площадке. Данное требование применимо в том случае, если указанный риск невозможно в достаточной степени контролировать с помощью организационных или технических мер. При классификации действующего вещества или лекарственного средства, обладающего высоким сенсибилизирующим потенциалом, необходимо учитывать, приводит ли его применение к высокой частоте сенсибилизации у человека или вероятность возникновения высокой частоты сенсибилизации у человека основана на данных, полученных с использованием животных, либо в результате других валидированных испытаний, а также учитывать тяжесть реакций гиперчувствительности, включая их в оценку весомости доказательств отнесения действующего вещества или лекарственного средства к этой группе.
3. Терапевтические макромолекулы и пептиды
40. Терапевтические макромолекулы и пептиды подвержены деградации и денатурации под воздействием экстремальных значений pH и (или) нагревания и способны терять фармакологическую активность. Очистка биофармацевтического производственного оборудования выполняется в условиях, при которых поверхности оборудования подвержены воздействию экстремальных значений pH и (или) высоких температур, что приводит к деградации и инактивации белковых препаратов. В связи с этим может не потребоваться установление пределов воздействия на здоровье с использованием PDE для сохраняющего активность действующего вещества.
41. При наличии других потенциальных путей перекрестной контаминации возникающие риски следует анализировать отдельно для каждого случая.
4. Недостаточность данных о репродуктивной
и онтогенетической токсичности
42. В целях обеспечения защиты всех популяций, подвергающихся экспозиции действующего вещества (контаминанта), присутствие остаточного действующего вещества (контаминанта) необходимо сократить до значения, которое не будет представлять риск влияния на репродуктивные и онтогенетические параметры. Вместе с тем на ранней фазе разработки доклинические данные для оценки потенциала нового действующего вещества вызывать репродуктивную и онтогенетическую токсичность могут быть еще не получены. Также могут отсутствовать научные сведения о репродуктивной и онтогенетической токсичности зарегистрированных лекарственных препаратов (например, о потенциальной способности применяемых мужчинами лекарственных препаратов вызывать нежелательное влияние на эмбрио-фетальное развитие потомства). В этом случае при достаточном обосновании для расчета PDE допускается использовать NOAEL, полученную при долгосрочном или среднесрочном исследовании с применением дополнительного поправочного коэффициента (например, равного 10). При наличии соответствующих результатов исследований репродуктивной и онтогенетической токсичности родственных действующим веществам (контаминантам) химических соединений, допускается использовать класс-специфичный профиль для выявления опасности неиспытанного контаминанта путем применения метода аналогий.
5. Исследуемые лекарственные препараты
43. В отношении исследуемых лекарственных препаратов, находящихся на ранней стадии разработки (I и II фазы), оценка PDE может быть затруднена в связи с ограниченностью данных. В таких случаях для установления допустимых пределов воздействия на здоровье при достаточном обосновании допускается прибегнуть к альтернативному подходу, аналогичному многоуровневым подходам на основе порога токсикологической угрозы используя деление на категории определенных значений по умолчанию (например, на основе низкой (высокой) прогнозируемой фармакологической активности, низкой (высокой) токсичности, генотоксичности (канцерогенности)).
44. Поскольку большинство предельных значений по умолчанию определяются продолжительностью экспозиции при многократном применении, иногда обоснованно повышенное предельное значение, если лекарственное средство производится на одном оборудовании с другим лекарственным средством, предназначенным для краткосрочных клинических исследований.
45. По мере получения новых фармакологических и токсикологических данных для установления допустимых пределов воздействия на здоровье необходимо рассчитывать специфичные для соединения предельные значения в соответствии с подходом, изложенным в разделе IV настоящего Руководства.
6. Представление данных о стратегии установления допустимой
ежедневной экспозиции (PDE)
46. Идентификация критических эффектов при установлении PDE, в соответствии с пунктами 11 - 34 настоящего Руководства должна быть основана на всестороннем поиске информации в научных источниках, включая руководства и статьи, а также электронные научные базы данных. Информация о стратегии поиска и его результатах должна быть представлена в досье производственных площадок. После анализа стратегии и результатов поиска специалистом в области токсикологии, фармакологии или химии компания обязана включить в досье производственных площадок информацию об обсуждении критических конечных точек, вызывающих опасение, и обоснование выбора конечных точек и дозы, которые будут использоваться для расчета PDE.
47. Ссылки публикации об опорных исследованиях с участием животных и исследованиях с участием человека, использованные для расчета PDE, должны позволять проследить информацию до оригинального источника публикации и должны быть подвергнуты анализу с точки зрения качества их планирования и выполнения (дизайн исследования, описание результатов, правильность документирования и т.д.). Стратегия расчета PDE должна содержать обоснование выбора поправочных коэффициентов, использованных для расчета PDE. В рамках фармацевтических инспекций в целях упрощения формирования обзора данных о стратегии установления PDE титульная страница каждого подготовленного документа о стратегии установления PDE должна содержать резюме обоснования стратегии установления допустимой ежедневной экспозиции (PDE), согласно приложению к настоящему Руководству.
Приложение
к Руководству
по установлению допустимых
пределов воздействия на здоровье
с целью идентификации рисков
при производстве различных
лекарственных средств
на общих производственных
(технологических) линиях
ФОРМА
резюме обоснования стратегии установления допустимой
ежедневной экспозиции (PDE)
Наименование юридического лица
Адрес места осуществления деятельности
Имя и подпись специалиста
Дата
Дата проведения оценки
Химическое название (названия) действующего вещества (контаминанта)
Идентифицированные опасности
ДА
НЕТ
НЕИЗВЕСТНО
Генотоксикант
Репродуктивный (онтогенетический) токсикант
Канцероген
Высокий сенсибилизирующий потенциал
Обоснование для расчета PDE
Обоснование выбора ведущего критического эффекта, используемого для расчета окончательного PDE, выбора NOAEL и выбора использованных поправочных коэффициентов, использованных для расчета PDE
Ссылка (ссылки) на публикацию (публикации), использованную при определении критического эффекта и дозы действующего вещества (контаминанта)
Резюме специалиста (сведения об образовании, квалификации и опыте работы специалиста, выполняющего или составившего обоснование стратегии установления допустимой ежедневной экспозиции (PDE))