Рекомендация Коллегии Евразийской экономической комиссии от 08.10.2019 N 29
КОЛЛЕГИЯ ЕВРАЗИЙСКОЙ ЭКОНОМИЧЕСКОЙ КОМИССИИ
РЕКОМЕНДАЦИЯ
от 8 октября 2019 г. N 29
О МЕТОДИЧЕСКИХ РЕКОМЕНДАЦИЯХ
ПО СОДЕРЖАНИЮ И СТРУКТУРЕ ДОКУМЕНТОВ РЕГИСТРАЦИОННОГО ДОСЬЕ
МЕДИЦИНСКОГО ИЗДЕЛИЯ
Коллегия Евразийской экономической комиссии в соответствии со статьей 31 Договора о Евразийском экономическом союзе от 29 мая 2014 года и статьей 4 Соглашения о единых принципах и правилах обращения медицинских изделий (изделий медицинского назначения и медицинской техники) в рамках Евразийского экономического союза от 23 декабря 2014 года,
в целях установления единообразных подходов при регистрации медицинских изделий к регистрационному досье, содержанию и структуре документов, содержащихся в регистрационном досье,
рекомендует государствам - членам Евразийского экономического союза с даты опубликования настоящей Рекомендации на официальном сайте Евразийского экономического союза применять Методические рекомендации по содержанию и структуре документов регистрационного досье медицинского изделия согласно приложению.
Председатель Коллегии
Евразийской экономической комиссии
Т.САРКИСЯН
Приложение
к Рекомендации Коллегии
Евразийской экономической комиссии
от 8 октября 2019 г. N 29
МЕТОДИЧЕСКИЕ РЕКОМЕНДАЦИИ
ПО СОДЕРЖАНИЮ И СТРУКТУРЕ ДОКУМЕНТОВ РЕГИСТРАЦИОННОГО ДОСЬЕ
МЕДИЦИНСКОГО ИЗДЕЛИЯ
I. Общие положения
1. Настоящие Методические рекомендации разработаны в целях оказания методической помощи заявителям при подготовке комплекта документов и материалов для регистрации медицинского изделия (далее - регистрационное досье) в соответствии с приложением N 4 к Правилам регистрации и экспертизы безопасности, качества и эффективности медицинских изделий, утвержденным Решением Совета Евразийской экономической комиссии от 12 февраля 2016 г. N 46 (далее - Правила регистрации).
Настоящие Методические рекомендации содержат разъяснения требований к содержанию и структуре документов регистрационного досье.
2. Для целей применения настоящих Методических рекомендаций под страной происхождения медицинского изделия понимается страна, в которой медицинское изделие было произведено или подвергнуто достаточной переработке в соответствии с критериями, определенными Правилами определения происхождения товаров, ввозимых на таможенную территорию Евразийского экономического союза (непреференциальными правилами определения происхождения товаров), утвержденными Решением Совета Евразийской экономической комиссии от 13 июля 2018 г. N 49.
Иные понятия, используемые в настоящих Методических рекомендациях, применяются в значениях, определенных актами органов Евразийского экономического союза (далее - Союз) в сфере обращения медицинских изделий.
II. Требования к содержанию и структуре документов
регистрационного досье медицинского изделия
3. Для экспертизы и регистрации медицинского изделия заявитель предоставляет (направляет) в уполномоченный орган (экспертную организацию) референтного государства следующие документы:
1) регистрационное досье на электронном носителе. В случае если законодательством государства - члена Союза (далее - государство-член) не предусмотрена возможность оформления указанных документов в электронном виде, уполномоченный орган референтного государства вправе запросить такие документы (их копии) на бумажном носителе. При этом документы, представленные на иностранном языке, должны иметь заверенный в установленном законодательством референтного государства порядке аутентичный перевод на русский язык.
Информация о соответствии сведений, содержащихся в регистрационном досье, структуре данных о медицинском изделии технического файла приведена в приложении к Методическим рекомендациям по проведению экспертизы безопасности, качества и эффективности медицинских изделий в целях их регистрации в рамках Евразийского экономического союза (приложение к Рекомендации Коллегии Евразийской экономической комиссии от 21 мая 2019 г. N 14);
2) копии документов, подтверждающих оплату экспертизы и регистрации медицинского изделия в референтном государстве.
Заявитель несет расходы на экспертизу и регистрацию медицинского изделия в соответствии с законодательством референтного государства, на согласование экспертного заключения в соответствии с законодательством государств признания.
4. Документы, указанные в пункте 3 настоящих Методических рекомендаций, предоставляются заявителем в уполномоченный орган (экспертную организацию) референтного государства на бумажном носителе непосредственно или заказным почтовым отправлением с уведомлением о вручении и описью вложения либо в электронной форме, в том числе на электронном носителе, согласно Требованиям к электронному виду заявлений и документов регистрационного досье, представляемых при осуществлении регистрации и экспертизы безопасности, качества и эффективности медицинских изделий, утвержденным Решением Коллегии Евразийской экономической комиссии от 30 июня 2017 г. N 78.
Заявителю необходимо обеспечить полноту, достоверность, наглядность и доступность предоставляемой информации, а также оперативность ее предоставления.
Ответственность за достоверность информации и сведений, содержащихся в предоставленном в уполномоченный орган (экспертную организацию) регистрационном досье, несет заявитель.
5. Документы, указанные в пункте 3 настоящих Методических рекомендаций, принимаются уполномоченным органом (экспертной организацией) референтного государства по описи, копия которой с отметкой о дате приема указанных документов вручается заявителю или направляется ему заказным почтовым отправлением с уведомлением о вручении или в электронном виде.
6. В составе регистрационного досье заявитель предоставляет следующие документы, предусмотренные приложением N 4 к Правилам регистрации:
1) заявление - заявления о проведении экспертизы и регистрации медицинского изделия по формам согласно приложениям N 2 и 3 к Правилам регистрации, которые заполняются и заверяются на официальном бланке (в случае наличия такого бланка) заявителя.
В сведениях о производителе (пункт 12 формы заявления о проведении экспертизы и пункт 8 формы заявления о проведении регистрации, предусмотренных приложениями N 2 и 3 к Правилам регистрации соответственно) страна определяется с учетом места регистрации производителя, от имени которого медицинское изделие выпускается в обращение на таможенной территории Союза.
При указании сведений о производственной площадке (производственных площадках) (пункт 13 формы заявления о проведении экспертизы и пункт 9 формы заявления о проведении регистрации, предусмотренных приложениями N 2 и 3 к Правилам регистрации соответственно) следует руководствоваться понятием "производственная площадка", установленным абзацем девятнадцатым пункта 3 Правил регистрации. При этом под определенными стадиями производственного процесса понимаются:
изготовление всего медицинского изделия или его основных блоков, кроме основных блоков, являющихся медицинскими изделиями, зарегистрированными в установленном порядке в Союзе, в территориально обособленных комплексах, входящих в организационную структуру производителя медицинского изделия;
изготовление по договорам со сторонним производителем (подрядчиком) всего медицинского изделия или его основных блоков, кроме основных блоков, являющихся медицинскими изделиями, зарегистрированными в установленном порядке в Союзе;
стерилизация медицинского изделия;
2) доверенность от производителя на право представления интересов при регистрации (при необходимости) - доверенность оформляется в соответствии с законодательством референтного государства. Для уполномоченного представителя производителя доверенность должна наделять его правом представлять интересы производителя, в том числе, при регистрации медицинских изделий, а также в части ответственности за обращение медицинского изделия в рамках Союза и исполнения обязательных требований, предъявляемых к медицинским изделиям;
3) копия разрешительного документа на право производства в стране-производителе с приложением (при наличии) - выданный уполномоченным органом документ с указанием номера и даты выдачи, дающий право осуществлять соответствующий вид деятельности, содержащий сведения об адресах мест осуществления соответствующего вида деятельности (при наличии, в случае законодательно установленного требования в стране производителя);
4) копии сертификатов на систему менеджмента качества производителя медицинских изделий (ИСО 13485 либо соответствующий региональный или национальный стандарт государства-члена) (при наличии) - документы, выданные на имя производителя (производственной площадки), подтверждающие внедрение производителем (производственной площадкой) системы менеджмента качества при производстве медицинских изделий в соответствии с требованиями стандартов, эквивалентных международному стандарту ISO 13485;
5) декларация о соответствии требованиям безопасности и эффективности медицинских изделий или эквивалентный документ (при наличии) - документ либо его копия, подтверждающий соответствие медицинского изделия обязательным требованиям государств, не являющихся членами Союза (например, директивам или регламентам Европейского союза);
6) копия регистрационного удостоверения (сертификата свободной продажи, сертификата на экспорт (за исключением медицинских изделий, впервые произведенных на территории государства-члена)), выданного в стране производителя (при наличии) с представлением перевода на русский язык;
7) копия документа, удостоверяющего регистрацию в других странах (при наличии) - предоставляется для информации о том, что медицинское изделие зарегистрировано на территории других государств. Могут быть предоставлены сведения о регистрации в других странах со ссылкой на источники таких сведений (например, ссылка на базу данных, другие массивы информации, содержащиеся в информационных системах);
8) справка на медицинское изделие с описанием области применения, назначения, краткой характеристики медицинского изделия, вариантами исполнения и комплектующими (по форме) - справка составляется по форме, предусмотренной приложением N 4 к Правилам регистрации, в которой описываются краткие данные о медицинском изделии, в том числе указывается комплектность медицинского изделия с разделением на основные блоки (части), принадлежности, расходные материалы, комплектующие в соответствии с Критериями разграничения элементов медицинского изделия, являющихся составными частями медицинского изделия, в целях его регистрации, утвержденными Решением Коллегии Евразийской экономической комиссии от 24 июля 2018 г. N 116;
9) данные о маркировке и упаковке (полноцветные макеты упаковок и этикеток, текст маркировки на русском языке и государственных языках государств-членов) - данные предоставляются в соответствии с требованиями к маркировке медицинского изделия согласно пунктам 58 - 63 и 105 Общих требований безопасности и эффективности медицинских изделий, требований к их маркировке и эксплуатационной документации на них, утвержденных Решением Совета Евразийской экономической комиссии от 12 февраля 2016 г. N 27 (далее - Общие требования). Также могут быть представлены фотографические изображения общего вида медицинского изделия и принадлежностей, необходимых для его применения по назначению;
10) информация о разработке и производстве: схемы процессов производства, основные стадии производства, упаковка, испытания и процедура выпуска конечного продукта - информация, представленная производителем в свободной форме, в которой описывается производственный процесс (возможно, в виде схем) с указанием производственной площадки (производственных площадок), а также страна происхождения, которая определяется с учетом места происхождения основного (основных) блоков медицинского изделия в соответствии с Правилами определения происхождения товаров, ввозимых на таможенную территорию Евразийского экономического союза (непреференциальными правилами определения происхождения товаров);
11) сведения о производителе: наименование, вид деятельности, юридический адрес, форма собственности, состав руководства, перечень подразделений и дочерних компаний с указанием их статуса и полномочий - в качестве таких сведений могут быть представлены копии документа о регистрации юридического лица в соответствии с законодательством страны производителя, устава организации и т.д.;
12) информация о маркетинге (история при условии обращения изделия на рынке более 2 лет) (при наличии) - (предоставляется для медицинского изделия 2б и 3 классов потенциального риска применения) информация, представляемая в свободной форме с указанием сведений о времени выхода медицинского изделия на рынок, о рынках, на которых обращается медицинское изделие, а также ориентировочного суммарного количества выпущенных медицинских изделий;
13) сообщения о несчастных случаях и отзывах (информация не предоставляется для вновь разработанных и спроектированных медицинских изделий): список нежелательных событий или несчастных случаев, связанных с использованием изделия, и указание периода времени, на протяжении которого происходили указанные случаи, если нежелательных событий слишком много, необходимо предоставить краткие обзоры по каждому из типов событий и указать общее количество событий каждого типа, о которых поступали отчеты, список отзывов с рынка медицинских изделий и (или) пояснительных уведомлений и описание подхода к рассмотрению этих проблем и их решению производителями в каждом из таких случаев, описание анализа и (или) корректирующих действий, предпринятых в ответ на указанные случаи - (не представляются для медицинского изделия для диагностики in vitro 1 класса потенциального риска применения) документ о неблагоприятных событиях (инцидентах), произошедших на всех этапах обращения медицинских изделий в рамках Союза и на территориях государств, не являющихся членами Союза, составленный производителем в свободной форме;
14) перечень стандартов, которым соответствует медицинское изделие (с указанием сведений о них) - документ, в котором указываются обозначения и наименования стандартов, включенных в перечень стандартов, в результате применения которых на добровольной основе полностью или частично обеспечивается соблюдение соответствия медицинских изделий Общим требованиям безопасности и эффективности медицинских изделий, требованиям к их маркировке и эксплуатационной документации на них (приложение к Рекомендации Коллегии Евразийской экономической комиссии от 4 сентября 2017 г. N 17), либо иных стандартов, которые применяются для доказательства соответствия медицинского изделия Общим требованиям. В случае если стандарт применяется не полностью в сведениях о таком стандарте указываются его структурные элементы;
15) сведения о соответствии медицинского изделия общим требованиям безопасности и эффективности медицинских изделий, требованиям к их маркировке и эксплуатационной документации на них - сведения, представляемые по форме в соответствии с приложением N 2 к Общим требованиям. Доказательные материалы (документы), в том числе протоколы испытаний (исследований), информация о собственных методах (методиках) испытаний приводятся в качестве приложений к указанной форме или как самостоятельные документы досье;
16) документ, устанавливающий требования к техническим характеристикам медицинского изделия - документ, составленный производителем, содержащий основные характеристики медицинского изделия (например, технические условия, стандарты организации, спецификации);
17) протоколы технических испытаний, проведенных в целях доказательства соответствия Общим требованиям - (не предоставляются для реагентов, наборов реагентов) протоколы испытаний медицинского изделия, выданные организациями, включенными в единый реестр уполномоченных организаций, имеющих право проводить исследования (испытания) медицинских изделий в целях их регистрации, оформленные в соответствии с приложением к Правилам проведения технических испытаний медицинских изделий, утвержденным Решением Совета Евразийской экономической комиссии от 12 февраля 2016 г. N 28;
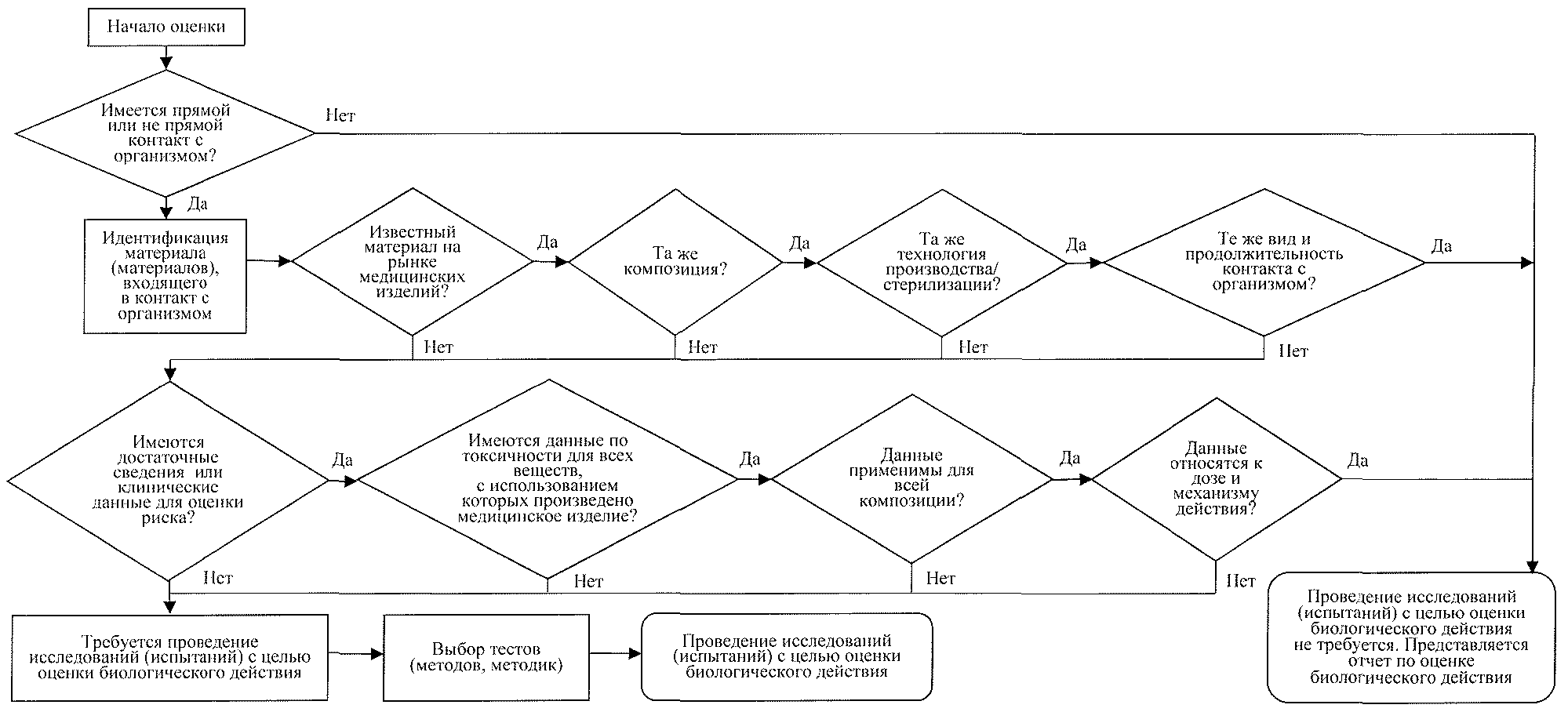
18) протоколы исследований (испытаний) по оценке биологического действия медицинского изделия, проведенных в целях доказательства соответствия Общим требованиям - протоколы исследований (испытаний) медицинского изделия, выданные организациями, включенными в единый реестр уполномоченных организаций, имеющих право проводить исследования (испытания) медицинских изделий в целях их регистрации, оформленные в соответствии с приложением к Правилам проведения исследований (испытаний) с целью оценки биологического действия медицинских изделий, утвержденным Решением Совета Евразийской экономической комиссии от 16 мая 2016 г. N 38.
При оценке биологического действия медицинского изделия рекомендуется использовать алгоритм планирования проведения исследований (испытаний) с целью оценки биологического действия медицинского изделия согласно приложению N 1.
В случаях, когда имеющихся сведений о материалах медицинского изделия и (или) принадлежностей к медицинским изделиям, контактирующих с поверхностью тела человека, его слизистыми оболочками, внутренними средами организма, достаточно для доказательства соответствия Общим требованиям, проведение исследований (испытаний) с целью оценки биологического действия медицинских изделий не требуется. При этом заявитель в составе досье представляет в уполномоченный орган (экспертную организацию) референтного государства отчет по оценке биологического действия, являющийся частью отчета об анализе рисков и содержащий сведения, обосновывающие отсутствие необходимости проведения исследований (испытаний) с целью оценки биологического действия;
19) отчет о клиническом доказательстве эффективности и безопасности медицинского изделия - (не предоставляется для медицинского изделия для диагностики in vitro 1 класса потенциального риска применения) должен содержать анализ и оценку всех имеющихся клинических данных (как благоприятных, так и неблагоприятных), на основании которых делается заключение о клинической безопасности и эффективности в соответствии с требованиями, установленными в Правилах проведения клинических и клинико-лабораторных испытаний (исследований) медицинских изделий, утвержденных Решением Совета Евразийской экономической комиссии от 12 февраля 2016 г. N 29. Отчет о клиническом доказательстве эффективности и безопасности медицинского изделия должен поддерживаться в актуальном состоянии с учетом данных, полученных в ходе постпродажного мониторинга и (или) при появлении новой подтвержденной информации из научных литературных источников на основании опыта клинического применения медицинского изделия, касающейся его безопасности и эффективности, и содержать данные обо всех выявленных неблагоприятных событиях (инцидентах) и противопоказаниях. Актуальная версия отчета предоставляется в уполномоченный орган (экспертную организацию) референтного государства по запросу.
Сведения, указанные в инструкции по применению медицинского изделия, должны соответствовать данным, содержащимся в отчете о клиническом доказательстве эффективности и безопасности медицинского изделия. В случае если медицинское изделие применялось только в отношении определенных групп пациентов, это должно быть указано в отчете о клиническом доказательстве эффективности и безопасности медицинского изделия и инструкции по применению медицинского изделия.
Отчет о клиническом доказательстве эффективности и безопасности медицинского изделия рекомендуется согласовывать с медицинским специалистом в области медицинского применения медицинского изделия. Квалификация медицинского специалиста подтверждается документом об образовании в области медицины и о специальности в соответствии с областью медицинского применения медицинского изделия (сертификатом специалиста, дипломом и т.д.), публикациями медицинского специалиста (научными статьями, монографиями, диссертациями в области медицинского применения медицинского изделия).
Рекомендованная структура отчета о клиническом доказательстве эффективности и безопасности медицинского изделия приведена в Методических рекомендациях по проведению экспертизы безопасности, качества и эффективности медицинских изделий в целях их регистрации в рамках Евразийского экономического союза (приложение к Рекомендации Коллегии Евразийской экономической комиссии от 21 мая 2019 г. N 14).
Сведения в отчете о клиническом доказательстве эффективности и безопасности медицинского изделия представляются в соответствии с указаниями согласно приложению N 2;
20) отчет об анализе рисков - (не предоставляется для медицинского изделия 1 класса потенциального риска применения) документ, составленный производителем, в котором идентифицированы все качественные и количественные характеристики, которые могут повлиять на безопасность медицинского изделия, определены опасности, связанные с применением медицинского изделия, как в нормальных условиях функционирования, так и в условиях отказа в работе или иного события, которое может привести к опасным ситуациям и причинению вреда жизни, здоровью человека, окружающей среде, и оценены связанные с ними риски. В соответствии с пунктом 4 Общих требований производитель должен показать, что он оценивает риски и управляет ими так, чтобы остаточные риски были допустимыми.
Для конкретных идентифицированных производителем опасностей и опасных ситуаций, которые определены стандартами по безопасности медицинских изделий, указанными в перечне стандартов, в результате применения которых на добровольной основе полностью или частично обеспечивается соблюдение соответствия медицинских изделий Общим требованиям безопасности и эффективности медицинских изделий, требованиям к их маркировке и эксплуатационной документации на них (приложение к Рекомендации Коллегии Евразийской экономической комиссии от 4 сентября 2017 г. N 17), производитель может не определять или не оценивать соответствующие риски. В этом случае выполнение требований стандартов может считаться валидированной мерой по управлению риском.
Для доказательства соответствия медицинского изделия положениям раздела II Общих требований производители медицинских изделий 1 класса потенциального риска применения могут ссылаться на имеющийся отчет об анализе риска;
21) данные о лекарственных средствах в составе медицинского изделия (состав лекарственного средства, количество, данные о совместимости лекарственного средства с медицинским изделием, регистрации лекарственного средства в стране-производителе) - сведения, указанные в технической документации производителя, включающие дату и номер регистрации лекарственного средства, входящего в состав медицинского изделия, в стране-производителе лекарственного средства. Данные о совместимости лекарственного средства с медицинским изделием могут быть приведены путем ссылки на соответствующие документы досье;
22) данные о биологической безопасности (при наличии) - сведения о материалах животного или человеческого происхождения, наночастицах, генно-модифицированных организмах и других вновь разрабатываемых материалах, входящих в медицинское изделие, а также информация о подборе источников (доноров), отборе материала, процессинге, хранении, тестировании, валидации процедур тестирования, обращения с тканями, клетками, субстанциями животного или человеческого происхождения, культурами микроорганизмов и вирусов;
23) данные о процедуре стерилизации, включая информацию о валидации процесса, результаты тестирования на содержание микроорганизмов (степень биологической нагрузки), пирогенности, стерильности (при необходимости) с указанием методов проведения испытаний и данные о валидации упаковки (для стерильных изделий) - (не предоставляется для медицинского изделия для диагностики in vitro 1 класса потенциального риска применения) документ, составленный производителем в соответствии с требованиями нормативных документов, применимых к регистрируемому медицинскому изделию (например, стандартов);
24) информация о специальном программном обеспечении (при наличии): сведения производителя о валидации программного обеспечения - документ, составленный производителем в соответствии с требованиями нормативных документов, применимых к регистрируемому медицинскому изделию (например, стандартов);
25) отчет об исследованиях стабильности (с аутентичным переводом на русский язык результатов и выводов испытаний для изделий, имеющих срок хранения) - документ, составленный производителем в соответствии с требованиями нормативных документов, применимых к регистрируемому медицинскому изделию (например, стандартов), подтверждающий способность медицинского изделия сохранять химические, физические свойства в определенных границах на протяжении срока годности (с указанием температурного режима, влажности, количества образцов, партии, даты их изготовления). Содержит сведения, необходимые для подтверждения стабильности изделия, а также его составных частей (например, реагентов, картриджей) во время их планового применения (в реальных или искусственно созданных условиях). Возможно включение данных о стабильности открытых флаконов и (или) стабильности реагентов, помещенных в прибор. К информации, которую рекомендуется включить в данный отчет, относятся:
обоснование выбора проведенных исследований;
все полученные результаты проведенных исследований, анализ данных, обоснование критериев приемлемости;
обоснование того, что безопасность или эффективность медицинского изделия не меняются в установленных документацией производителя условиях хранения.
Для медицинских изделий, для которых не устанавливается срок годности (например, электромедицинского оборудования или других изделий, пригодных для многоразового использования), может быть представлена информация относительно оценки среднего срока службы или отчеты о стабильности методом ускоренного старения. Информация о сроке службы может быть представлена, например, сведениями о количестве процедур, которое можно выполнить с помощью данного медицинского изделия в определенный период времени;
26) эксплуатационный документ или инструкция по применению медицинского изделия на государственных языках государств признания (при необходимости) и русском языке - документ, составленный производителем содержащий информацию в соответствии с Общими требованиями который может включать в себя в том числе руководство по эксплуатации, методику медицинского применения, паспорт, формуляр, инструкции по монтажу, наладке, техническому обслуживанию, ремонту, транспортировке, хранению, утилизации медицинского изделия;
27) руководство по сервисному обслуживанию (в части комплектующих медицинского изделия) - в случае отсутствия данных в эксплуатационной документации (при наличии) - документ, составленный производителем, регламентирующий порядок сервисного обслуживания медицинского изделия, при необходимости дополняющий эксплуатационный документ или инструкцию по применению медицинского изделия;
28) отчет об инспекции производства (при наличии) - документ, составленный инспектирующей организацией в соответствии с Требованиями к внедрению, поддержанию и оценке системы менеджмента качества медицинских изделий в зависимости от потенциального риска их применения, утвержденными Решением Совета Евразийской экономической комиссии от 10 ноября 2017 г. N 106;
29) план сбора и анализа данных по безопасности и эффективности медицинских изделий на постпродажном этапе - документ, составленный производителем для медицинских изделий всех классов потенциального риска применения. План сбора и анализа данных по безопасности и эффективности медицинских изделий на постпродажном этапе может предусматривать такие способы получения данных, как, например, отзывы потребителей об опыте применения медицинского изделия, анкетирование пользователей, записи результатов устных опросов и другие формы и отчеты в соответствии с документированной процедурой обратной связи с потребителем, предусмотренной системой менеджмента качества предприятия.
В плане сбора и анализа данных по безопасности и эффективности медицинских изделий на постпродажном этапе также указываются сроки проведения всех мероприятий.
План сбора и анализа данных по безопасности и эффективности медицинских изделий для диагностики in vitro на постпродажном этапе применим для подтверждения их аналитической и клинической эффективности;
30) документы, подтверждающие результаты испытаний медицинских изделий в целях утверждения типа средств измерений (в отношении медицинских изделий, включенных в перечень видов медицинских изделий, подлежащих отнесению при их регистрации к средствам измерений, утвержденный Решением Совета Евразийской экономической комиссии от 12 февраля 2016 г. N 42) (при необходимости) - документы со сведениями о результатах проведения испытаний, выданные организациями, уполномоченными (нотифицированными) в соответствии с законодательством государства-члена на проведение испытаний средств измерений.
7. Документы, указанные в подпунктах 2 - 4 и 6 пункта 6 настоящих Методических рекомендаций, заверяются в соответствии с международными нормами заверения или нормами заверения, установленными в соответствии с законодательством референтного государства.
В странах - участницах Конвенции, отменяющей требование легализации иностранных официальных документов, от 5 октября 1961 года, на документах должен быть проставлен апостиль, в странах, не являющихся участниками указанной конвенции - документы должны быть легализованы через консульские службы (нотариально заверены), в странах - участницах Конвенции о правовой помощи и правовых отношениях по гражданским, семейным и уголовным делам от 22 января 1993 года - нотариально заверены.
8. Документы, указанные в подпунктах 1, 7 - 16, 19 - 27 и 29 пункта 6 настоящих Методических рекомендаций, заверяются производителем (его уполномоченным представителем).
9. При предоставлении документов регистрационного досье в электронном виде (либо в виде электронных копий документов) они должны соответствовать следующим требованиям:
а) документы, указанные в подпунктах 1, 7 - 16, 19 - 29 пункта 6 настоящих Методических рекомендаций, должны быть представлены в формате *.pdf с текстовым слоем с возможностью выделения и копирования блоков, а также осуществления поиска;
б) документы, указанные в подпунктах 2 - 6, 17, 18 и 30 пункта 6 настоящих Методических рекомендаций, должны быть представлены в формате *.pdf с разрешением не менее 300 dpi;
в) размер любого из файлов регистрационного досье не должен превышать 100 Мб согласно Правилам электронного обмена данными в интегрированной информационной системе внешней и взаимной торговли, утвержденным Решением Коллегии Евразийской экономической комиссии от 27 января 2015 г. N 5. В случае превышения указанного размера допускается разделение документа на несколько файлов;
г) все документы регистрационного досье обязательно должны иметь реквизит "дата выдачи документа".
Приложение N 1
к Методическим рекомендациям
по содержанию и структуре
документов регистрационного
досье медицинского изделия
АЛГОРИТМ
ПЛАНИРОВАНИЯ ПРОВЕДЕНИЯ ИССЛЕДОВАНИЙ (ИСПЫТАНИЙ) С ЦЕЛЬЮ
ОЦЕНКИ БИОЛОГИЧЕСКОГО ДЕЙСТВИЯ МЕДИЦИНСКОГО ИЗДЕЛИЯ
Приложение N 2
к Методическим рекомендациям
по содержанию и структуре
документов регистрационного
досье медицинского изделия
УКАЗАНИЯ
ПО ПРЕДСТАВЛЕНИЮ СВЕДЕНИЙ В ОТЧЕТЕ О КЛИНИЧЕСКОМ
ДОКАЗАТЕЛЬСТВЕ ЭФФЕКТИВНОСТИ И БЕЗОПАСНОСТИ
МЕДИЦИНСКОГО ИЗДЕЛИЯ
I. Общие положения
1. Отчет о клиническом доказательстве эффективности и безопасности медицинского изделия должен содержать информацию о клинической безопасности и эффективности медицинского изделия, основанную на всех имеющихся клинических данных о медицинском изделии, и может состоять из нескольких разделов (в случае, если клинические данные получены разными методами).
Производитель самостоятельно определяет перечень разделов отчета о клиническом доказательстве эффективности и безопасности медицинского изделия исходя из примененных им методов получения клинических данных.
2. В клинических данных о медицинском изделии должны быть указаны сроки, цели и задачи проведения клинических либо клинико-лабораторных испытаний (исследований), группы пациентов, критерии выбора пациентов, выявленные в ходе клинических либо клинико-лабораторных испытаний (исследований) неблагоприятные события (инциденты), вид проведенных испытаний (например, испытания, проведенные в целях регистрации либо проведенные на пострегистрационном этапе на территориях государств, не являющихся членами Евразийского экономического союза (далее - Союз)), полученные статистические данные клинических либо клинико-лабораторных испытаний (исследований), материалы и методы, которые были использованы в ходе клинических либо клинико-лабораторных испытаний (исследований), наименования медицинских организаций, в которых проводились клинические либо клинико-лабораторные испытания (исследования), и другая информация в соответствии с международными требованиями по надлежащей клинической практике.
3. К клиническим данным о медицинском изделии, полученным в результате проведения клинических либо клинико-лабораторных испытаний (исследований) в Союзе в соответствии с Правилами проведения клинических и клинико-лабораторных испытаний (исследований) медицинских изделий, утвержденными Решением Совета Евразийской экономической комиссии от 12 февраля 2016 г. N 29, должны прилагаться программа клинического либо клинико-лабораторного испытания (исследования) и отчет о клиническом либо клинико-лабораторном испытании (исследовании) медицинского изделия.
4. Отзывы медицинских организаций об опыте применения медицинского изделия без предоставления клинических данных о медицинском изделии в соответствии с международными требованиями не могут быть признаны в качестве доказательства эффективности и безопасности медицинского изделия.
II. Клинические данные о медицинском изделии, полученные
при проведении клинических испытаний (исследований)
медицинского изделия или при применении медицинского
изделия в государствах, не являющихся членами Союза
5. Клинические данные о медицинском изделии, полученные при проведении клинических испытаний (исследований) или при применении медицинского изделия в государствах, не являющихся членами Союза, могут быть представлены в отчете о клиническом доказательстве эффективности и безопасности медицинского изделия для доказательства эффективности и безопасности медицинского изделия в случае, если такие данные подтверждают эффективность и безопасность медицинского изделия по показаниям к медицинскому применению в соответствии с назначением медицинского изделия. Клинические данные о медицинском изделии, полученные при проведении клинических испытаний (исследований) в государствах, не являющихся членами Союза, предоставляемые заявителем в отчете о клиническом доказательстве эффективности и безопасности медицинского изделия, признаются уполномоченным органом (экспертной организацией) в качестве источника клинических данных о медицинском изделии в случае выполнения одного из следующих условий:
а) клинические данные подтверждены публикациями в специализированных журналах или отчетами Всемирной организации здравоохранения по программе контроля безопасности и эффективности медицинских изделий ("The WHO prequalification project"), размещенными на сайте Всемирной организации здравоохранения в информационно-телекоммуникационной сети "Интернет".
К специализированным журналам относятся любые журналы международного уровня, специализирующиеся в области медицинского применения регистрируемого медицинского изделия, а также публикации на международных веб-ресурсах и в базах данных в области здравоохранения (например, Medscape, Medline, Pubmed, new England Journal of Medicine). Публикации должны относиться к регистрируемому медицинскому изделию и позволять оценивать его. Предоставление публикаций о технологии применения медицинского изделия (например, о проведении стентирования, радиологической терапии и т.д.) вместо публикаций об оценке конкретного регистрируемого медицинского изделия недопустимо;
б) представлены результаты проведения клинических испытаний (исследований) медицинского изделия в соответствии с рекомендациями Международного форума регуляторов медицинских изделий (IMDRF). Проведенные клинические испытания (исследования) медицинского изделия должны позволять верифицировать их соответствие международным требованиям.
III. Клинические данные о медицинском изделии,
эквивалентном регистрируемому медицинскому изделию
6. Клинические данные о другом медицинском изделии могут быть приняты к рассмотрению уполномоченным органом (экспертной организацией) в качестве доказательства эффективности и безопасности регистрируемого изделия только при представлении заявителем доказательств его эквивалентности регистрируемому медицинскому изделию путем одновременного выполнения следующих условий:
а) рассматриваемые медицинские изделия имеют одинаковое назначение;
б) технические и биологические характеристики рассматриваемых медицинских изделий являются одинаковыми в той степени, которая гарантирует отсутствие различий в их клинической эффективности и безопасности.
7. Эквивалентное медицинское изделие, клинические данные о котором используются для обоснования клинической безопасности и эффективности регистрируемого медицинского изделия, может быть не зарегистрировано в государствах - членах Союза (далее - государство-член). При этом для такого эквивалентого медицинского изделия предоставляется копия регистрационного удостоверения (сертификата свободной продажи, сертификата на экспорт), выданного в стране производителя, с представлением перевода на русский язык.
Требования к объему и степени детализации клинических данных, полученных для эквивалентных медицинских изделий, те же, что и для клинических данных регистрируемого медицинского изделия.
8. Клинические данные о медицинском изделии, эквивалентном регистрируемому медицинскому изделию, должны соответствовать установленным в рамках Союза требованиям к клиническим данным о медицинском изделии.
9. Клинические данные о медицинском изделии, эквивалентном регистрируемому медицинскому изделию, полученные при проведении клинических испытаний (исследований) в государствах, не являющихся членами Союза, должны соответствовать требованиям, установленным разделом II настоящего документа.
IV. Клинические данные о медицинских изделиях классов
потенциального риска применения 3, 2б и имплантируемых
медицинских изделиях (за исключением медицинских
изделий для диагностики in vitro)
10. Обоснование клинической эффективности и безопасности медицинских изделий классов потенциального риска применения 3, 2б и имплантируемых медицинских изделий должно основываться на клинических данных о медицинском изделии, полученных при проведении многоцентровых клинических испытаний (исследований).
Результаты клинических испытаний (исследований), которые были проведены до 1 января 2016 г. (по дате последнего визита последнего пациента или субъекта испытания (исследования)) или продолжали выполняться по состоянию на 1 января 2016 г. (при завершенном наборе пациентов) на территории государства-члена, могут быть приняты к рассмотрению уполномоченным органом (экспертной организацией) при условии, что такие клинические испытания (исследования) были проведены в соответствии с законодательством этого государства-члена. При этом медицинские организации, в которых проводились клинические испытания (исследования), должны соответствовать требованиям законодательства государства-члена, на территории которого проводились клинические испытания (исследования), и необязательно должны быть включены в единый реестр уполномоченных организаций, имеющих право проводить исследования (испытания) медицинских изделий в целях их регистрации. Результаты таких клинических испытаний (исследований) должны быть оформлены в виде отчета о клиническом испытании (исследовании) медицинского изделия в соответствии с законодательством государства-члена, на территории которого они проводились, и утверждены координатором-исследователем.
Результаты клинических испытаний (исследований), которые были проведены до 1 января 2016 г. (по дате последнего визита последнего пациента или субъекта испытания (исследования)) или продолжали выполняться по состоянию на 1 января 2016 г. (при завершенном наборе пациентов) на территориях государств, не являющихся членами Союза, могут быть приняты к рассмотрению уполномоченным органом (экспертной организацией) при условии, что такие клинические испытания (исследования) были проведены в соответствии с рекомендациями Международного форума регуляторов медицинских изделий (IMDRF) и утверждены координатором-исследователем.
Результаты клинических испытаний (исследований), инициированных после 1 января 2016 г., могут быть приняты к рассмотрению уполномоченным органом (экспертной организацией) при условии, что клинические испытания (исследования) были проведены в соответствии с Правилами проведения клинических и клинико-лабораторных испытаний (исследований) медицинских изделий, утвержденными Решением Совета Евразийской экономической комиссии от 12 февраля 2016 г. N 29, при этом одно из клинических испытаний (исследований) должно быть проведено в одном из государств-членов.
11. Для медицинских изделий класса потенциального риска применения 3, 2б и имплантируемых медицинских изделий в отчет о клиническом доказательстве эффективности и безопасности медицинского изделия включается план пострегистрационного клинического мониторинга в соответствии с Правилами проведения мониторинга безопасности, качества и эффективности медицинских изделий, утвержденными Решением Коллегии Евразийской экономической комиссии от 22 декабря 2015 г. N 174 (далее - Правила проведения мониторинга).
План пострегистрационного клинического мониторинга должен содержать информацию и сведения, установленные пунктом 21 Правил проведения мониторинга.
В плане пострегистрационного клинического мониторинга также указываются сроки его проведения. Отчеты о пострегистрационном клиническом мониторинге безопасности и эффективности медицинских изделий, предоставляемые в соответствии с Правилами проведения мониторинга, оцениваются уполномоченным органом (экспертной организацией) также на предмет их соответствия плану пострегистрационного клинического мониторинга. При необходимости в план пострегистрационного клинического мониторинга могут вноситься изменения в рамках актуализации данных, содержащихся в регистрационном досье.
V. Клинические данные о медицинских изделиях
для диагностики in vitro
12. В отношении медицинских изделий для диагностики in vitro не является обязательным:
проведение многоцентровых клинико-лабораторных испытаний (исследований);
в случае проведения многоцентровых клинико-лабораторных испытаний (исследований) - проводить их в одном из государств-членов.
Производитель вправе проводить многоцентровые клинико-лабораторные испытания (исследования) медицинских изделий для диагностики in vitro.
13. Для доказательства эффективности и безопасности медицинских изделий для диагностики in vitro классов потенциального риска применения 2б и 3 достаточно провести клинико-лабораторные испытания (исследования) в одной медицинской организации.