"МР 2.6.1.0064-12. 2.6.1. Ионизирующее излучение, радиационная безопасность. Радиационный контроль питьевой воды методами радиохимического анализа. Методические рекомендации"
ФЕДЕРАЛЬНАЯ СЛУЖБА ПО НАДЗОРУ В СФЕРЕ ЗАЩИТЫ
ПРАВ ПОТРЕБИТЕЛЕЙ И БЛАГОПОЛУЧИЯ ЧЕЛОВЕКА
ГЛАВНЫЙ ГОСУДАРСТВЕННЫЙ САНИТАРНЫЙ ВРАЧ
РОССИЙСКОЙ ФЕДЕРАЦИИ
Утверждаю
Руководитель Федеральной службы
по надзору в сфере защиты
прав потребителей и
благополучия человека,
Главный государственный
санитарный врач
Российской Федерации
Г.Г.ОНИЩЕНКО
9 июня 2012 г.
Дата введения: с момента утверждения
2.6.1. ИОНИЗИРУЮЩЕЕ ИЗЛУЧЕНИЕ. РАДИАЦИОННАЯ БЕЗОПАСНОСТЬ
РАДИАЦИОННЫЙ КОНТРОЛЬ
ПИТЬЕВОЙ ВОДЫ МЕТОДАМИ РАДИОХИМИЧЕСКОГО АНАЛИЗА
МЕТОДИЧЕСКИЕ РЕКОМЕНДАЦИИ
МР 2.6.1.0064-12
1. Авторский коллектив: Федеральное бюджетное учреждение науки "Санкт-Петербургский научно-исследовательский институт радиационной гигиены имени профессора П.В. Рамзаева" Роспотребнадзора
(М.В. Кадука, В.Н. Шутов, Н.С. Швыдко, Л.Н. Басалаева, Н.В. Салазкина, Ю.Н. Гончарова, О.С. Кравцова, И.П. Стамат, В.В. Ступина).
2. Утверждены Руководителем Федеральной службы по надзору в сфере защиты прав потребителей и благополучия человека, Главным государственным санитарным врачом Российской Федерации.
Г.Г. Онищенко "9" июня 2012 г.
3. Введены в действие с момента утверждения.
4. Введены впервые.
Список сокращений, принятых в настоящих
методических рекомендациях
НКДАР ООН
- научный комитет по действию атомной радиации при
организации объединенных наций;
ВОЗ
- всемирная организация здравоохранения;
УВ
- уровень вмешательства;
св.
- свыше;
п.
- пункт;
УМФ
- установка малофоновая;
![]() -излучающие
-излучающие
- альфа-излучающие;
![]() -излучающие
-излучающие
- бета-излучающие;
отн. ед.
- относительные единицы;
ОСК
- образец сравнения контрольный;
х.ч.
- химически чистый;
ч.д.а.
- чистый для анализа;
ДПР
- дочерние продукты распада;
МВИ
- методика выполнения измерений;
НД
- нормативный документ;
КУ
- контрольный уровень
1. ОБЛАСТЬ ПРИМЕНЕНИЯ
1.1. Настоящие Методические рекомендации (далее - Рекомендации) распространяются на порядок приготовления счетных образцов для определения удельной суммарной альфа- и бета-активности и удельной активности основных дозообразующих радионуклидов в питьевой воде и выполнение контроля качества питьевой воды по показателям радиационной безопасности.
1.2. Основной целью Рекомендаций является обеспечение единых подходов при выполнении анализов удельной суммарной альфа- и бета-активности, а также радионуклидного состава и удельной активности основных природных (исключая 222Rn) и техногенных радионуклидов в воде.
1.3. Настоящие Рекомендации предназначены для органов и организаций Роспотребнадзора в субъектах Российской Федерации, осуществляющих контроль и санитарно-эпидемиологическую оценку питьевой воды по показателям радиационной безопасности, а также аккредитованных в установленном порядке на право проведения соответствующих измерений лабораторий радиационного контроля и организаций, обеспечивающих централизованное водоснабжение населения при проведении производственного радиационного контроля.
2. НОРМАТИВНЫЕ ССЫЛКИ
2.1. Федеральный закон от 09.01.96 N 3-ФЗ "О радиационной безопасности населения".
2.2. Федеральный закон от 30.03.99 N 52-ФЗ "О санитарно-эпидемиологическом благополучии населения".
2.3. СанПиН 2.6.1.2523-09 "Нормы радиационной безопасности (НРБ-99/2009)".
2.4. СП 2.6.1.2612-10 "Основные санитарные правила обеспечения радиационной безопасности (ОСПОРБ-99/2010)".
2.5. СанПиН 2.6.1.2800-10 "Гигиенические требования по ограничению облучения населения за счет природных источников ионизирующего излучения".
2.6. МУ 2.6.1.1981-05 "Радиационный контроль и гигиеническая оценка источников питьевого водоснабжения и питьевой воды по показателям радиационной безопасности. Оптимизация защитных мероприятий источников питьевого водоснабжения с повышенным содержанием радионуклидов".
2.7. МУ 2.6.1.2719-10 "Изменение 1 к МУ 2.6.1.1981-05 "Радиационный контроль и гигиеническая оценка источников питьевого водоснабжения и питьевой воды по показателям радиационной безопасности. Оптимизация защитных мероприятий источников питьевого водоснабжения с повышенным содержанием радионуклидов".
2.8. ГОСТ Р 51592-2000 "Вода. Общие требования к отбору проб".
2.9. СанПиН 2.1.4.1074-01 "Питьевая вода. Гигиенические требования к качеству воды централизованных систем питьевого водоснабжения. Контроль качества".
2.10. Письмо Федеральной службы по надзору в сфере защиты прав потребителей и благополучия человека от 21.08.2006 N 0100/9009-06-32 "О радиационном контроле за питьевой и минеральной водой".
2.11. МР 0100/13609-07-34 "Отбор и подготовка проб питьевой воды для определения показателей радиационной безопасности".
2.12. ГОСТ Р 8.594-2002 "Метрологическое обеспечение радиационного контроля".
3. ТЕРМИНЫ И ОПРЕДЕЛЕНИЯ
В настоящем документе принята терминология в соответствии с НРБ-99/2009 и ОСПОРБ-99/2010. В дополнение к ним используются следующие термины:
Контрольный источник - радионуклидный источник ионизирующего излучения, предназначенный для калибровки средств измерений ионизирующих излучений.
Контрольный образец сравнения - радиоактивный источник излучения, служащий для калибровки по нему других источников ионизирующих излучений, утвержденный в качестве образцового в установленном порядке.
Коэффициент связи - коэффициент, связывающий активность счетного образца со скоростью счета импульсов от его излучения.
Минимальная измеряемая активность - Амин - активность радионуклида в счетном образце, при измерении которой на данной радиометрической установке за время экспозиции один час относительная случайная (статистическая) погрешность результата измерений составляет 50% при доверительной вероятности P = 0,95.
Носитель - вещество, которое, будучи связано с ничтожно малым (индикаторным) количеством другого вещества, проносит последнее через весь химический или физический процесс.
Проба воды - определенное количество воды, отобранное из контролируемого источника в соответствии с принятой методикой отбора проб.
Радиометрическая установка - техническое средство (радиометр, спектрометр) для измерения активности (удельной активности) радионуклидов в счетном образце.
Счетный образец - определенное количество вещества, полученное из пробы согласно установленной методике и предназначенное для измерений его параметров на радиометрической установке в соответствии с регламентированной методикой выполнения измерений.
Суммарная активность - условная активность счетного образца, численно равная активности регламентированного образца сравнения при одинаковых показаниях радиометра.
Химический выход радионуклида - отношение количества носителя радионуклида в измеряемом образце к количеству носителя этого радионуклида, добавленного в пробу.
4. ОБЩИЕ ПОЛОЖЕНИЯ
Природные радионуклиды можно разделить на две большие группы - первичные, т.е. те, которые образовались одновременно со стабильным веществом Земли, и космогенные, которые образуются постоянно в результате ядерных реакций под действием космического излучения или поступают со внеземным веществом. Очевидно, к настоящему моменту в окружающей среде присутствуют только те первичные радионуклиды, период полураспада которых соизмерим с возрастом Земли [1].
Три первичных радионуклида - 238U, 235U и 232Th - являются родоначальниками естественных радиоактивных рядов (Приложение 1). С течением времени в естественных радиоактивных рядах установилось вековое равновесие - состояние, в котором активности (но не количества ядер) всех членов ряда равны между собой. Вековое равновесие между радионуклидами устанавливается в том случае, если период полураспада материнского радионуклида велик по сравнению с периодом полураспада дочернего. Для установления радиоактивного равновесия достаточен промежуток времени, равный 10 периодам полураспада дочернего радионуклида. Различные геохимические процессы могут приводить к фракционированию членов радиоактивных рядов, поэтому вековое равновесие сохраняется только в системе, замкнутой относительно материнского и дочерних радионуклидов.
По данным НКДАР ООН, вклад питьевой воды в суммарную дозу облучения населения не является преобладающим (за исключением отдельных регионов) и обусловлен, в основном, присутствующими в воде радионуклидами природных рядов урана и тория. Наибольший вклад в формирование дозы облучения за счет потребления питьевой воды вносят изотопы урана (238U и 234U), радия (226Ra и 228Ra), радон (222Rn) и полоний-210 (210Po), в меньшей степени - свинец-210 (210Pb) и изотопы тория (228Th, 230Th, 232Th) (МУ 2.6.1.1981-05).
Для обеспечения радиационной безопасности питьевого водоснабжения населения необходима информация об удельной активности радионуклидов, присутствующих в питьевой воде. В соответствии с рекомендациями ВОЗ в действующих нормативных документах установлены жесткие требования к содержанию радионуклидов и к порядку проведения радиационного контроля питьевой воды.
Проведение радиационного контроля воды является сложной задачей, поскольку сама вода является сложным объектом для анализа.
В силу чрезвычайной вариабельности радионуклидного состава и удельной активности природных радионуклидов, минерального состава и других характеристик природных вод необходимо использование, по возможности, прямых радиохимических методов определения содержания радионуклидов.
Следует подчеркнуть, что большинство известных радиохимических методик определения природных радионуклидов предусматривает их анализ из одной пробы воды, из которой отбираются отдельные аликвотные части от 0,5 до 2,0 кг для определения удельной активности групп изотопов одного или двух элементов. Вместе с тем, информация, получаемая из одной пробы при последовательном выделении из нее радиоизотопов урана, радия, свинца и полония, более достоверна, особенно учитывая высокую степень вариабельности радионуклидного состава природных вод. Реализация этого подхода нуждается в серьезных радиохимических проработках по созданию схем последовательного разделения и идентификации различных нуклидов из одной пробы, что, помимо указанных выше преимуществ, позволяет сделать процесс более экономичным - сокращает время анализа, расход реактивов, энерго- и трудозатраты.
Настоящая методика обеспечивает определение суммарных радиологических показателей воды и удельной активности природных (226Ra, 224Ra, 228Ra, 210Po, 210Pb, 210Bi, 238U, 234U, 232Th, 228Th, 230Th, 40K) и техногенных радионуклидов (137Cs, 90Sr) из одной пробы воды, из которой отбираются аликвоты: 1 кг для определения удельной суммарной альфа- и бета-активности, 10 кг - для определения удельной активности природных радионуклидов и не менее 4 кг для определения удельной активности 137Cs, 90Sr и 40K (4 кг на техногенно загрязненных территориях и 20 кг для определения удельной активности радионуклидов на уровне фоновых значений).
Радиационный контроль воды проводится в несколько этапов:
- отбор и доставка проб для анализа;
- предварительная оценка соответствия питьевой воды критериям радиационной безопасности (определение удельной суммарной альфа- и бета-активности);
- исследование радионуклидного состава воды (при превышении контрольного уровня);
- оценка качества воды по радиологическим показателям.
Для получения достоверной оценки радиологических показателей воды на каждом этапе контроля необходимо выполнять определенные требования.
На первом этапе, при отборе проб воды (Приложение 2 Рекомендаций), должны использоваться емкости из полимерных материалов для исключения сорбции микроколичеств радионуклидов. Важно также, чтобы отобранная вода была подкислена (HCl) и своевременно подверглась процедуре пробоподготовки - срок хранения пробы перед определением суммарных показателей и удельных активностей природных радионуклидов не должен превышать 14 дней (при необходимости определения удельной активности 222Rn срок хранения не должен превышать 2 дней). Нарушение указанных процедур может привести к неконтролируемым потерям радионуклидов, а значит, к искажению результатов. В акте отбора проб (Приложение 3 Рекомендаций) должна содержаться вся информация, необходимая для идентификации источника водоснабжения.
На втором этапе измеряется удельная суммарная альфа- и бета-активность воды для предварительной оценки соответствия питьевой воды критериям радиационной безопасности. Для питьевой воды подземных источников водоснабжения одновременно с измерением удельной ![]() - и
- и ![]() -активности требуется определять содержание 222Rn. Время от отбора до доставки пробы воды в лабораторию с целью определения удельной активности 222Rn должно быть не более двух суток.
-активности требуется определять содержание 222Rn. Время от отбора до доставки пробы воды в лабораторию с целью определения удельной активности 222Rn должно быть не более двух суток.
При превышении контрольных уровней удельной суммарной ![]() - и
- и ![]() -активности выполнение радионуклидного анализа воды обязательно, причем желательно с привлечением радиохимических методов, позволяющих дать точную количественную характеристику того или иного радионуклида в исследуемой пробе. Несмотря на сложность и трудоемкость радиохимических методов, необходимость привлечения специалистов высокой квалификации, преимущество использования радиохимических приемов для контроля и гигиенической оценки питьевой воды по показателям радиационной безопасности очевидно.
-активности выполнение радионуклидного анализа воды обязательно, причем желательно с привлечением радиохимических методов, позволяющих дать точную количественную характеристику того или иного радионуклида в исследуемой пробе. Несмотря на сложность и трудоемкость радиохимических методов, необходимость привлечения специалистов высокой квалификации, преимущество использования радиохимических приемов для контроля и гигиенической оценки питьевой воды по показателям радиационной безопасности очевидно.
Радиохимический анализ позволяет с высокой степенью надежности выделять радиохимически чистые изотопы, избегать косвенных оценок содержания радионуклидов, обладает высокой чувствительностью, точностью и информативностью.
При оценке радиационной безопасности питьевого водоснабжения населения радиохимический анализ питьевой воды должен обеспечить определение основных дозообразующих природных радионуклидов с обязательным повторным определением удельной суммарной активности ![]() - и
- и ![]() -излучающих радионуклидов из объема водной пробы, представленной для радиохимического анализа, для подтверждения соответствия суммарной альфа- и бета-активности и суммарной активности выделенных радионуклидов.
-излучающих радионуклидов из объема водной пробы, представленной для радиохимического анализа, для подтверждения соответствия суммарной альфа- и бета-активности и суммарной активности выделенных радионуклидов.
Радиохимическая подготовка, как правило, включает стадию концентрирования (при определении содержания природных радионуклидов в воде это выпаривание 10 кг до 1 кг), затем следует селективное выделение группы изотопов одного или нескольких элементов, отделение их от макрокомпонентов, радиохимическая очистка каждого радионуклида, приготовление счетного образца для измерения активности.
Проведение радионуклидного анализа воды с использованием 10 кг исходного материала позволяет обеспечивать минимально измеряемую удельную активность не более 0,2 от величины уровня вмешательства (УВ) для каждого радионуклида, что и требуется в нормативных документах, в частности в методических указаниях МУ 2.6.1.1981-05 "Радиационный контроль и гигиеническая оценка источников питьевого водоснабжения и питьевой воды по показателям радиационной безопасности. Оптимизация защитных мероприятий источников питьевого водоснабжения с повышенным содержанием радионуклидов".
5. ДИАПАЗОН ИЗМЕРЕНИЙ И ХАРАКТЕРИСТИКИ
ПОГРЕШНОСТИ ИЗМЕРЕНИЙ
5.1. Диапазон и относительная погрешность измерений радиологических показателей проб воды в предлагаемых Рекомендациях приведены в таблице 1.
Таблица 1
Диапазон и относительная погрешность измерений
радиологических показателей проб воды
Диапазон измерений, Бк/пробу
Относительная погрешность, % (P = 0,95)
238U, 234U, 230Th, 232Th, 228Th, 226Ra
210Pb, 228Ra, 224Ra
210Po, ![]()
40K, 137Cs, 90Sr, ![]()
св. 0,02 ![]() 0,05
0,05
включительно
50
50
50
50
св. 0,05 ![]() 0,10
0,10
включительно
40
40
40
50
св. 0,10 ![]() 0,50
0,50
включительно
30
30
30
40
св. 0,50 ![]() 1,00
1,00
включительно
20
20
20
30
св. 1,00 ![]() 5,00
5,00
включительно
20
20
20
20
свыше 5,00
20
20
20
20
Примечания к таблице 1:
1. Приведенные в таблице погрешности измерения величины радиологических показателей определены на нижней границе диапазона измерений для однократного измерения активности счетного образца, приготовленного из 1 кг пробы, и измерения собственного фона радиометра в течение не менее 1 часа для каждого.
2. При временах измерения активности счетного образца и собственного фона радиометра, отличных от 1 часа, погрешности измерения могут быть рассчитаны по формуле (25), приведенной в п. 13.11.3.
3. При использовании для приготовления счетного образца пробы массой более 1 кг границы диапазона измерений радиологических показателей проб воды в таблице 1, соответствующего данной относительной погрешности результатов измерений, необходимо поделить на величину, численно равную массе пробы воды в кг.
6. ПРИНЦИПЫ ОПРЕДЕЛЕНИЯ УДЕЛЬНОЙ СУММАРНОЙ
![]() - И
- И ![]() -АКТИВНОСТИ И УДЕЛЬНОЙ АКТИВНОСТИ
-АКТИВНОСТИ И УДЕЛЬНОЙ АКТИВНОСТИ
РАДИОНУКЛИДОВ В ПРОБАХ ВОДЫ
Из отобранной пробы воды выделяют аликвоты массой 1 кг для определения удельной суммарной ![]() - и
- и ![]() -активности, 10 кг - для определения удельной активности природных радионуклидов и не менее 4 кг для определения удельной активности 137Cs, 90Sr и 40K.
-активности, 10 кг - для определения удельной активности природных радионуклидов и не менее 4 кг для определения удельной активности 137Cs, 90Sr и 40K.
Для определения удельной суммарной ![]() - и
- и ![]() -активности <1> из аликвоты пробы массой 1 кг путем упаривания до сухого остатка приготавливают счетный образец, активность которого определяют на
-активности <1> из аликвоты пробы массой 1 кг путем упаривания до сухого остатка приготавливают счетный образец, активность которого определяют на ![]() -
-![]() радиометре, отградуированном в единицах активности.
радиометре, отградуированном в единицах активности.
--------------------------------
<1> В настоящем документе в качестве образца сравнения для измерения суммарной ![]() -активности используется хлорид калия с радионуклидом 40K. В качестве образца сравнения для суммарной
-активности используется хлорид калия с радионуклидом 40K. В качестве образца сравнения для суммарной ![]() -активности используется образец, изготовленный на основе 226Ra.
-активности используется образец, изготовленный на основе 226Ra.
Для определения удельной активности природных радионуклидов из аликвоты пробы массой 10 кг путем радиохимического анализа приготавливают счетные образцы, содержащие отдельные радионуклиды. Активность каждого радионуклида определяют с помощью ![]() -
-![]() радиометра и
радиометра и ![]() -спектрометра, предварительно отградуированных в единицах активности.
-спектрометра, предварительно отградуированных в единицах активности.
Определение удельной активности 137Cs и 90Sr проводят не ранее чем через 15 дней после отбора пробы воды, когда наступит радиоактивное равновесие между 90Sr и его дочерним продуктом распада - 90Y. Из аликвоты пробы массой не менее 4 кг 137Cs выделяют сурьмяно-иодидным методом, активность 90Sr определяют по активности его дочернего радионуклида 90Y, который осаждают оксалатным методом. Активность приготовленных счетных образцов определяют на ![]() -
-![]() радиометре УМФ-2000, который предварительно градуируют в единицах активности 137Cs и 90Y соответственно.
радиометре УМФ-2000, который предварительно градуируют в единицах активности 137Cs и 90Y соответственно.
Для контроля неизбежных в процессе анализа потерь радионуклидов в исследуемую пробу на самом первом этапе ее обработки для каждого из определяемых изотопов добавляют известное количество носителя, в качестве которого используют солянокислые или азотнокислые соли соответствующих элементов. После проведения анализа определяют количество оставшегося носителя. Отношение количества носителя, определенного на выходе, к количеству носителя, добавленного в пробу, дает величину химического выхода для каждого радионуклида:
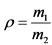 , (1)
, (1)
где: ![]() - химический выход изотопа, отн. ед.;
- химический выход изотопа, отн. ед.;
m1 - количество носителя, определенного на выходе, мг;
m2 - количество носителя, добавленного в пробу, мг.
7. СРЕДСТВА ИЗМЕРЕНИЙ И ВСПОМОГАТЕЛЬНОЕ ОБОРУДОВАНИЕ
7.1. Для выполнения измерений по настоящим Рекомендациям применяются следующие средства измерений:
7.1.1. ![]() -спектрометр с характеристиками не хуже следующих:
-спектрометр с характеристиками не хуже следующих:
- время установления рабочего режима - не более 10 мин.;
- интегральная нелинейность в диапазоне энергий 4 ![]() 9 МэВ - не более 0,1%;
9 МэВ - не более 0,1%;
- энергетическая ширина канала - не более 5,0 кэВ;
- эффективность регистрации излучения радионуклида 239Pu не менее 0,25 Бк-1·с-1 на расстоянии 5 мм от детектора;
- временная нестабильность характеристики преобразования - не более 2% за 8 часов измерений;
- абсолютное энергетическое разрешение по энергии 5,16 МэВ - менее 50 кэВ; 5,49 МэВ - менее 50 кэВ; 5,81 МэВ - менее 50 кэВ.
7.1.2. ![]() -
-![]() радиометр типа УМФ-2000 или другой с характеристиками не хуже следующих:
радиометр типа УМФ-2000 или другой с характеристиками не хуже следующих:
- скорость счета фона - не более 0,05 имп./мин. по ![]() -каналу и не более 2,3 имп./мин. - по
-каналу и не более 2,3 имп./мин. - по ![]() -каналу;
-каналу;
- эффективность регистрации ![]() -излучения 137Cs - не ниже 3,7 имп./(мин.·Бк) и 90Sr (90Y) - не ниже 7,5 имп./(мин.·Бк), соответственно <1>;
-излучения 137Cs - не ниже 3,7 имп./(мин.·Бк) и 90Sr (90Y) - не ниже 7,5 имп./(мин.·Бк), соответственно <1>;
--------------------------------
<1> Установлена для счетных образцов толщиной не более 0,06 г/см2.
- эффективность регистрации ![]() -излучения электрохимических счетных образцов с диаметром активного пятна 26 мм - не ниже 16,2 имп./(мин.·Бк);
-излучения электрохимических счетных образцов с диаметром активного пятна 26 мм - не ниже 16,2 имп./(мин.·Бк);
- основная погрешность определения активности счетных образцов - не более 10% при P = 0,95.
7.1.3. Эталонные радионуклидные источники и растворы радионуклидов:
- образцовые спектрометрические источники альфа-излучения 2-го разряда с радионуклидами 239Pu, 242Pu и 238Pu активностью в диапазоне 10 - 100 Бк;
- образцовый источник ![]() -излучения 2-го разряда с радионуклидом 90Sr (90Y) активностью в диапазоне 10 - 100 Бк;
-излучения 2-го разряда с радионуклидом 90Sr (90Y) активностью в диапазоне 10 - 100 Бк;
- эталонные растворы радионуклидов 226Ra, 228Ra, 232U, 210Pb, 137Cs, (90Sr + 90Y) объемной активностью от 10 до 50 Бк/см3 с погрешностью измерений объемной активности не более 6%;
- контрольный образец сравнения ОСК-210 (210Po + 210Bi), активностью от 10 до 50 Бк с погрешностью измерений активности не более 6%.
7.1.4. Средства измерения массы и объема, применяемые при приготовлении счетных образцов:
- весы аналитические АДВ-200, ГОСТ 24104-80;
- весы технические ВРЛ-1, ГОСТ 19491-74;
- набор разновесов, ГОСТ 7328-82;
- пипетки мерные 2-го класса на 1,5 и 10 мл, ГОСТ 20292-74;
- колбы мерные 2-го класса на 100, 250 и 1000 мл, ГОСТ 1770-78;
- стаканы лабораторные на 50, 200, 250, 300, 800, 1000 и 2000 мл, ГОСТ 10394-72.
7.2. Вспомогательное оборудование:
- плитка электрическая, ГОСТ 14919-83;
- дистиллятор, Д-4 ТУ 64-1-1640-78;
- печь муфельная электрическая типа ПМ-8 с терморегулятором до 900 °C;
- колонки хроматографические стеклянные с внутренним диаметром 10 мм;
- форвакуумный насос (ВН-461, ВН-2М), обеспечивающий рабочее давление в вакуумной камере не более 1333 Па (10 мм рт. ст.);
- разборная установка для электролитического осаждения радионуклидов на подложки из нержавеющей стали диаметром 34 мм, включающая электролитическую ячейку с анодным и катодным электродами;
- регулируемый источник питания постоянного тока, обеспечивающий максимальный ток до 3 А и максимальное напряжение до 30 В;
- никелевые диски диаметром 34 мм и толщиной 0,6 мм, изготовленные из никелевой ленты марки НП-2 (0,6 x 250) ГОСТ 2170-73;
- наждачная бумага мелкозерновая;
- индикаторная бумага универсальная pH 1 - 10, ТУ МХПОРУ 76-56;
- набор ареометров;
- стандартные сита;
- чашки и тигли фарфоровые на 25, 50 и 100 мл, ГОСТ Т 9147-80;
- чашки кварцевые на 50 мл, ГОСТ 10973-64;
- ступка фарфоровая;
- стекло часовое;
- палочки стеклянные диаметром 3 мм;
- шпатель;
- воронки стеклянные диаметром 5, 7,5, 10 и 15 см, ГОСТ 8613-75;
- колба круглодонная К-1-2000-29/32, ГОСТ 25336-82;
- колба коническая Кн-2-1000-34, ТУ 92-891.029-91;
- холодильник прямой ХПТ-1-600-14/23-14/23, ГОСТ 25336-82;
- подложки из нержавеющей стали марок 1Х18Н9Т, 12Х18Н10Т, 08Х18Н10Т или аналогичных ГОСТ 4543, ГОСТ 5632, диаметром 34 мм, толщиной 0,7 - 1,0 мм;
- алюминиевая фольга 81 г/см2, толщина 3 мм марок А5, А6, А7, АДО, АД31 ГОСТ 15175-89;
- тефлоновая кассета для электролитического осаждения;
- бумага фильтровальная, ГОСТ 12026-66;
- инфракрасная лампа мощностью 250 - 500 Вт;
- перчатки резиновые;
- ветошь, вата.
7.3. Для выполнения измерений по настоящим Рекомендациям могут применяться другие средства измерений и вспомогательное оборудование, характеристики которых не хуже перечисленных в п. 7.1.
7.4. Все средства измерений должны иметь действующие свидетельства о метрологической поверке.
8. РЕАКТИВЫ И МАТЕРИАЛЫ
Для выполнения измерений по настоящим Рекомендациям требуются следующие реактивы и материалы:
- аммиак водный NH4OH, ГОСТ 3760-79;
- аммоний иодистый NH4I, ГОСТ 3764-75;
- аммоний углекислый (NH4)2CO3, ГОСТ 3770-75;
- аммоний хлористый NH4Cl ГОСТ 3773-72;
- аммоний щавелевокислый (NH4)2C2O4, ГОСТ 5712-78;
- ацетон CH3COCH3, ГОСТ 2603-79;
- барий хлористый BaCl2, ГОСТ 41-08-72;
- вода дистиллированная H2O, ГОСТ 6709-72;
- железо хлорное 6-водное FeCl3·6H2O, ГОСТ 4147-74;
- иттрий хлористый 6-водный YCl3·6H2O, ТУ 6-09-4773-84;
- калий сернокислый K2SO4, ГОСТ 4145-74;
- калий хлористый KCl, ГОСТ 3760-79;
- кальций хлористый CaCl2, ТУ 6-09-02-401-86;
- карбонат натрия (сода) Na2CO3, ГОСТ 83-79;
- кислота азотная концентрированная HNO3, ГОСТ 44621-77;
- кислота аскорбиновая, НД 42-10043-99;
- кислота лимонная 1-водная, ГОСТ 3652-69;
- кислота серная концентрированная H2SO4, ГОСТ 4204-77;
- кислота соляная концентрированная HCl, ГОСТ 14261-77;
- кислота уксусная CH3COOH, ГОСТ 61-75;
- кислота щавелевая H2C2O4, ГОСТ 22180-76;
- лантан хлористый 7-водный LaCl3·7H2O, ТУ 6-09-4773-79;
- натрий сернистокислый Na2SO3, ГОСТ 195-77;
- натрий ферроцианид Na4Fe(CN)6, ГОСТ 4207-75;
- никель азотнокислый Ni(NO3)2, ГОСТ 4055-78;
- перекись водорода 30% H2O2, ГОСТ 10929-76;
- спирт этиловый C2H5OH, ГОСТ 17299-78;
- стронций хлористый 6-водный SrCl2·6H2O, ГОСТ 4140-74;
- сурьма треххлористая SbCl3, ТУ 6-09-626-76;
- трилон Б Na4-EDTA ТУ 2484-007-22657427-2000;
- уранил азотнокислый UO2(NO3)2·6H2O;
- цезий азотнокислый, CsNO3, ТУ 6-09-437-83;
- анионообменная смола ЭДЭ-10П, размер зерен 0,125 - 0,250 мм, ГОСТ 20301-74;
- катионообменная смола К-2-8, размер зерен 0,125 - 0,250 мм, ГОСТ 1305.
Примечание:
Все используемые для выполнения анализа по настоящим Рекомендациям реактивы и материалы должны быть марок х.ч. или ч.д.а. Процедура приготовления рабочих растворов и растворов носителей приведена в Приложении 4. Процедура приготовления калибровочных образцов и градуировки альфа-бета радиометра приведена в Приложении 5.
9. ТРЕБОВАНИЯ БЕЗОПАСНОСТИ
Работы по настоящим Рекомендациям должны проводиться с соблюдением требований:
- Инструкции по технике безопасности "Работа с вредными веществами";
- Норм радиационной безопасности НРБ-99/2009;
- Основных санитарных правил обеспечения радиационной безопасности ОСПОРБ-99/2010.
При работе с измерительными приборами и вспомогательным оборудованием необходимо соблюдать требования безопасной эксплуатации, изложенные в соответствующих разделах технической документации на приборы.
10. ТРЕБОВАНИЯ К КВАЛИФИКАЦИИ
К выполнению работ по настоящим Рекомендациям допускаются лица не моложе 18 лет, прошедшие обучение и практическую подготовку по соответствующей программе, имеющие квалификацию химика, оператора (лаборанта) по радиометрическим и спектрометрическим измерениям, врача по санитарно-гигиеническим лабораторным исследованиям, врача-лаборанта, фельдшера-лаборанта, или инженерно-технические работники, допущенные к выполнению работ в установленном порядке, ознакомленные с руководством по эксплуатации и техническим описанием ![]() -
-![]() радиометра и/или
радиометра и/или ![]() -спектрометра.
-спектрометра.
11. ПРИГОТОВЛЕНИЕ СЧЕТНЫХ ОБРАЗЦОВ ДЛЯ ИЗМЕРЕНИЯ УДЕЛЬНОЙ
СУММАРНОЙ АЛЬФА- И БЕТА-АКТИВНОСТИ ВОДЫ
11.1. При определении удельной суммарной альфа- и бета-активности необходимо соблюдать, по крайней мере, два условия:
- при прокаливании остатка, полученного в результате упаривания воды и ее сульфатации (для обеспечения идентичности матриц стандарта и пробы), температура не должна превышать 300 - 400 °C;
- следует соблюдать временной интервал измерения полученного счетного образца после его приготовления. При наличии в пробе воды 226Ra при прокаливании из препарата удаляется 222Rn, через 3 - 4 часа распадаются его дочерние продукты распада (ДПР) и именно по прошествии этого времени выполняют измерение суммарной активности ![]() и
и ![]() излучающих радионуклидов, поскольку в дальнейшем становится заметным накопление 222Rn и его ДПР из 226Ra, содержащегося в сухом остатке, что выражается в существенном увеличении скорости счета, особенно по
излучающих радионуклидов, поскольку в дальнейшем становится заметным накопление 222Rn и его ДПР из 226Ra, содержащегося в сухом остатке, что выражается в существенном увеличении скорости счета, особенно по ![]() -излучению, и результаты могут быть завышены в полтора-два раза. Если необходимо провести измерение образца еще раз, повторяют процедуру прокаливания и проводят измерения, соблюдая регламентированный временной интервал. При наличии в воде других изотопов радия, схемы изменения активности во времени усложняются, и для одной и той же пробы, в зависимости от времени отбора, приготовления образца и его измерения могут быть получены различающиеся данные. Особенности приготовления счетных образцов для проб воды стандартной минерализации, высокой минерализации и низкой минерализации приведены в пп. 11.2 - 11.4 Рекомендаций, соответственно.
-излучению, и результаты могут быть завышены в полтора-два раза. Если необходимо провести измерение образца еще раз, повторяют процедуру прокаливания и проводят измерения, соблюдая регламентированный временной интервал. При наличии в воде других изотопов радия, схемы изменения активности во времени усложняются, и для одной и той же пробы, в зависимости от времени отбора, приготовления образца и его измерения могут быть получены различающиеся данные. Особенности приготовления счетных образцов для проб воды стандартной минерализации, высокой минерализации и низкой минерализации приведены в пп. 11.2 - 11.4 Рекомендаций, соответственно.
В случае если измеренное значение удельной суммарной бета-активности пробы воды с учетом неопределенности измерений превышает 1,0 Бк/кг, проводят определение удельной активности 40K. Определение удельной активности 40K необходимо для корректной оценки соответствия питьевой воды требованиям радиационной безопасности. Присутствие в воде подземных водоисточников 40K обуславливает 50 - 90% суммарной величины бета-активности воды, при этом содержание данного природного радионуклида в питьевой воде не нормируется. При значении удельной суммарной бета-активности более 1,0 Бк/кг из данного измеренного значения вычитают значение удельной активности 40K в соответствии с санитарно-эпидемиологическими требованиями <1>. Оценку соответствия воды требованиям радиационной безопасности по удельной суммарной бета-активности проводят в данном случае на основании измеренного значения удельной суммарной бета-активности за вычетом удельной активности природного 40K. Определение удельной активности 40K проводят в соответствии с п. 13.10 Рекомендаций с использованием счетного образца, приготовленного для определения удельной суммарной альфа- и бета-активности пробы воды.
--------------------------------
<1> Пункт 4.3.6 СанПиН 2.6.1.2800-10 "Гигиенические требования по ограничению облучения населения за счет природных источников ионизирующего излучения", утвержденные постановлением Главного государственного санитарного врача Российской Федерации от 24.12.2010 N 171 (зарегистрировано Минюстом России 27.01.2011, регистрационный номер 19587).
11.2. При приготовлении счетных образцов для измерения удельной суммарной альфа- и бета-активности проб воды с минерализацией 100 - 600 мг/л выпаривают 1 кг воды до объема 100 мл в стеклянном огнеупорном стакане, переносят в фарфоровую выпарительную чашку, стакан обмывают 10 мл 10% H2SO4, 10 мл дистиллированной воды и переливают в ту же выпарительную чашку. Содержимое чашки выпаривают на электрической плитке до удаления паров H2SO4 и образования сухого остатка, который затем прокаливают в муфельной печи в течение 1 ч при температуре 300 - 400 °C. После охлаждения осадок растирают до состояния "пудры", взвешивают, аликвотную часть наносят на подложку. Рекомендуемая площадь распределения осадка 2,5 см2, затем осадок фиксируют этиловым спиртом и высушивают. Высушивание подготовленного счетного образца можно осуществлять под инфракрасной лампой.
11.3. Счетные образцы для измерения удельной суммарной альфа- и бета- активности проб воды с минерализацией свыше 600 мг/л приготавливают согласно п. 11.2 Рекомендаций.
При измерении счетного образца, приготовленного из проб воды с высокой степенью минерализации, на подложку наносится лишь малая аликвотная часть сухого остатка пробы, полученного после выпаривания и сульфатации, что существенно увеличивает погрешность определения значений удельной суммарной альфа- и бета-активности. В пробах воды с высокой минерализацией скорость счета радиометра от аликвотной (малой по весу по сравнению с полным весом) части образца, приготовленного для определения удельной суммарной альфа- и бета-активности, может отличаться от скорости счета фона самого радиометра на незначительную величину при использовании подложек площадью 2,5 см2, что приводит к увеличению погрешности проводимого измерения. Для увеличения значения счета радиометра по альфа- и бета-каналам и точности получаемого результата измерения используют подложки площадью 7 см2 или более для измерения суммарной альфа- и бета-активности счетных образцов, приготовленных из проб воды с минерализацией, превышающей 600 мг/л.
Удельную суммарную альфа- и бета-активность приготовленного счетного образца измеряют с использованием малофонового альфа-бета радиометра не ранее 3-х и не позднее 10 часов после последнего прокаливания (для исключения влияния 222Rn и ДПР) в соответствии с п. 13.2 Рекомендаций. У градуировочных и счетных образцов геометрия измерения и площадь подложки должны быть одинаковыми.
Измерение суммарной альфа- и бета-активности необходимо проводить не менее 3 - 4 раз, чередуя измерения счетного образца с измерением скорости счета фона радиометрической установки. Для расчета удельной суммарной альфа- и бета-активности усредняют измеренные значения суммарных показателей, исключая (в случае их наличия) значения, явно выбивающиеся из общего ряда измеренных, обусловленные, скачками фона радиометрической установки или случайными флуктуациями скорости счета радиометрической установки от счетного образца.
11.4. Счетные образцы для измерения удельной суммарной альфа- и бета- активности проб воды с минерализацией менее 100 мг/л приготавливают согласно общим принципам п. 11.2 Рекомендаций. При минерализации пробы воды менее 100 мг/л для достижения достаточной толщины и площади поверхности слоя счетного образца в выпарительную фарфоровую чашку с сухим остатком пробы, добавляют соль, не оказывающую влияние на скорость счета счетного образца (нейтральную соль), например, Ca3(PO4)2.
Фарфоровую чашку перед выпариванием пробы, предварительно взвешивают, вес чашки фиксируют в рабочем журнале. После упаривания до сухих солей и прокаливания в муфельной печи, фарфоровую чашку с пробой остужают и взвешивают повторно. По разнице веса чашки с образцом, полученным из пробы воды, и веса предварительно взвешенной пустой фарфоровой чашки, вычисляют вес сухого остатка пробы (минерализацию).
Если вес сухого остатка пробы воды составит менее 100 мг, в выпарительную фарфоровую чашку с сухим остатком пробы добавляют соль, не оказывающую влияние на активность счетного образца (нейтральную по активности соль), например, Ca3(PO4)2 до суммарного веса образца в чашке 100 мг (существующая геометрия, при которой проводилась калибровка радиометра). Смесь сухого остатка пробы с нейтральной солью растирают до состояния "пудры", наносят на подложку площадью 2,5 см2, фиксирует спиртом и высушивают.
Удельную суммарную альфа- и бета-активность приготовленного счетного образца измеряют с использованием малофонового альфа-бета радиометра не ранее 3-х и не позднее 10 часов после последнего прокаливания (для исключения влияния 222Rn и ДПР) в соответствии с п. 13.2 Рекомендаций.
12. РАДИОХИМИЧЕСКОЕ ВЫДЕЛЕНИЕ РАДИОНУКЛИДОВ
И ПРИГОТОВЛЕНИЕ СЧЕТНЫХ ОБРАЗЦОВ ДЛЯ ОПРЕДЕЛЕНИЯ
ИХ УДЕЛЬНОЙ АКТИВНОСТИ В ПРОБАХ ВОДЫ
Методика предусматривает осаждение изотопов радия из раствора пробы вместе с носителем (хлористый барий) серной кислотой в форме BaSO4, который является счетным образцом для измерения активности 226Ra и 224Ra.
228Ra определяют по дочернему 228Ac, который осаждают совместно с носителем (хлористый лантан) в виде La(OH)3. После последующего прокаливания полученный La2O3(228Ac) служит счетным образцом для измерения активности 228Ac(228Ra).
Радиоактивные изотопы полония, висмута (свинца), урана и тория соосаждают с гидроокисью железа с последующим их селективным выделением и определением:
- 210Po и 210Bi электролитически на никелевом диске, который является счетным образцом;
- 238U (при отсутствии в лаборатории ![]() -спектрометра) по дочернему 234Th, выделенному на носителе (хлористый иттрий) в виде оксалата, при прокаливании которого получают счетный образец Y2O3(234Th) для измерения активности определяемого радионуклида;
-спектрометра) по дочернему 234Th, выделенному на носителе (хлористый иттрий) в виде оксалата, при прокаливании которого получают счетный образец Y2O3(234Th) для измерения активности определяемого радионуклида;
- 238U, 234U, 228Th, 230Th, 232Th с применением ![]() -спектрометра. В этом случае для определения химического выхода урана и тория в качестве изотопных индикаторов используют стандартные растворы 232U и 234Th.
-спектрометра. В этом случае для определения химического выхода урана и тория в качестве изотопных индикаторов используют стандартные растворы 232U и 234Th.
Счетным образцом для определения удельной активности 137Cs является Cs3Sb2I9.
90Sr определяют по дочернему 90Y, для этого используют счетный образец Y2O3, полученный осаждением 90Y с носителем (хлористый иттрий) в виде оксалата и последующим прокаливанием.
Удельную активность 40K определяют радиометрически на основе измерений слоя половинного уменьшения его ![]() -излучения.
-излучения.
Схема радиохимического определения активности указанных радионуклидов в пробах воды представлена в Приложении 6.
12.1. Радиохимическое выделение 226Ra, 224Ra, 228Ac(228Ra)
12.1.1. Радий - радиоактивный элемент II группы периодической системы элементов. По химическим свойствам он весьма сходен с барием. Единственное валентное состояние равно 2+.
Слаборастворимые соли радия - сульфат и карбонат. Свойства солей радия в основном аналогичны свойствам солей бария, однако растворимость последних, как правило, несколько выше.
Три изотопа радия являются членами природных радиоактивных семейств: 226Ra (T1/2 = 1600 лет, ![]() -излучатель) образуется при
-излучатель) образуется при ![]() -распаде 230Th; 224Ra (T1/2 = 3,64 сут.) - продукт
-распаде 230Th; 224Ra (T1/2 = 3,64 сут.) - продукт ![]() -распада 228Th; 228Ra (T1/2 = 5,77 лет) образуется при
-распада 228Th; 228Ra (T1/2 = 5,77 лет) образуется при ![]() -распаде 232Th.
-распаде 232Th.
Определение изотопов радия основано на выделении суммы изотопов радия - 226Ra, 228Ra, 224Ra в виде сульфата радия-бария и непосредственном измерении ![]() -активности 226Ra и 224Ra в динамике.
-активности 226Ra и 224Ra в динамике.
Определение 228Ra основано на осаждении дочернего 228Ac (T1/2 = 6,13 ч) с носителем лантаном и измерении ![]() -активности выделенного препарата на
-активности выделенного препарата на ![]() -
-![]() радиометре (или
радиометре (или ![]() -радиометре).
-радиометре).
12.1.2. Солянокислый концентрат пробы воды (около 1 л), полученный после выпаривания 10 кг воды, в которую добавлена HCl до pH = 1, и содержащий в качестве носителя хлорид бария BaCl2 (90 - 100 мг в расчете на BaSO4) и хлорид железа FeCl3 (20 мг в расчете на Fe2O3), нагревают до кипения на электрической плитке в стакане емкостью 2 л, добавляют при непрерывном перемешивании 30 мл 10% H2SO4, кипятят 10 - 15 минут, после чего осадку дают отстояться в течение 3 - 4 часов (лучше оставить на ночь).
Прозрачный раствор над осадком сливают в стакан объемом 800 мл, осадок отфильтровывают в колбу объемом 500 мл, используя обеззоленный фильтр "синяя лента" диаметром 12,5 см (масса золы одного фильтра - 0,0004 г), несколько раз промывают дистиллированной водой (фильтрат 1). Время фильтрования записывают - это время отделения 224Ra от 228Th. Промытый осадок сульфата бария BaSO4 (осадок 1) переносят в стакан объемом 150 мл с помощью 70 - 100 мл дистиллированной воды, добавляют 5 - 7 г Na2CO3 и кипятят при периодическом перемешивании 2 часа, подливая время от времени дистиллированную воду, чтобы объем составлял около 100 мл. Осадок карбонатов Ba(Ra)CO3 отфильтровывают, используя обеззоленный фильтр "синяя лента" диаметром 12,5 (11,0) см (осадок 2), промывают 5% раствором Na2CO3 до отрицательной реакции на сульфат-ион, а затем один раз дистиллированной водой. Полученный фильтрат (фильтрат 2) подкисляют HCl до pH = 2, кипятят до удаления CO2 и объединяют с фильтратом 1.
Осадок Ba(Ra)CO3 (осадок 2) растворяют в 2 н HCl, кипятят до полного удаления углекислоты. В пробу вносят раствор хлорного железа (~10 мг на Fe2O3) и осаждают безугольным аммиаком Fe(OH)3 для дополнительной очистки радия от изотопов полония, висмута, свинца, тория и урана. Осадок (осадок 3) отфильтровывают, используя обеззоленный фильтр "красная лента" диаметром 12,5 (11,0) см (записывают время фильтрования), промывают горячей водой, растворяют в 2 н HCl и объединяют с фильтратом 1.
Фильтрат, полученный после отделения Fe(OH)3 и содержащий радиохимически чистые изотопы 224Ra, 226Ra, 228Ra, подкисляют HCl до pH = 2, вносят раствор носителя лантана (60 мг на La2O3) и оставляют на 36 - 40 часов для накопления 228Ac, затем раствор нагревают до температуры 70 °C и аммиаком (без углекислоты) осаждают гидроокись лантана (актиния). Осадок отфильтровывают, используя обеззоленный фильтр "синяя лента" диаметром 12,5 см (время окончания фильтрования, т.е. время отделения 228Ac от 228Ra, записывают), промывают горячей дистиллированной водой с добавлением безугольного аммиака, высушивают, прокаливают при температуре 800 °C, растирают в тигле стеклянной палочкой до состояния "пудры", переносят на подложку, фиксируют несколькими каплями этилового спирта, равномерно распределяют, высушивают под инфракрасной лампой и взвешивают для определения химического выхода.
Измерение активности 228Ac в приготовленном счетном образце и определение его удельной активности в пробе воды выполняют в соответствии с п. 13.3.
Фильтрат 3 (после отделения 228Ac на осадке гидроокиси лантана) подкисляют HCl до pH = 1, нагревают до кипения и осаждают сульфат бария (радия) 10% раствором H2SO4. После отстаивания осадок отфильтровывают, используя обеззоленный фильтр "синяя лента" диаметром 12,5 см, промывают дистиллированной водой, высушивают и прокаливают при температуре 800 °C в течение 1 часа. Записывают время окончания прокаливания. Осадок растирают, равномерно наносят на подложку и взвешивают на аналитических весах.
Измерение активности 224Ra и 226Ra в приготовленном счетном образце и определение их удельной активности в пробе воды выполняют в соответствии с п. 13.4.
12.2. Радиохимическое выделение 210Po и 210Bi(210Pb)
Метод определения полония основан на электрохимическом осаждении радионуклида на никелевом диске из солянокислых растворов. Этот метод называется электролитическим замещением или бестоковым электролизом.
Определение 210Pb производят по дочернему 210Bi, который так же, как и 210Po, электрохимически осаждается на никелевом диске.
При определении полония следует принимать меры для предотвращения его сорбции на стекле; растворы должны быть 0,7 н по соляной кислоте и не ниже 0,5 - 1% по лимонной кислоте.
Во всех случаях при осаждении полония на никелевый диск следует применять только соляную кислоту, а не азотную, т.к. последняя растворяет (особенно сильно при нагревании) никель.
Осаждению полония на никелевый диск мешает ион трехвалентного железа, если количество его в анализируемом растворе превышает 10 мг. Для устранения влияния железа добавляют аскорбиновую кислоту, восстанавливающую трехвалентное железо в двухвалентное, которое не мешает электрохимическому осаждению полония. Объем раствора не должен превышать 100 мл.
Методика выделения 210Po сводится к следующему: объединенные после осаждения сульфатов фильтраты (фильтрат 1, фильтрат 2 и раствор осадка 3) упаривают до 600 - 800 мл и аммиаком, не содержащим CO2, при pH = 9 осаждают гидрат окиси железа, с которым соосаждаются изотопы урана, тория, полония, свинца, висмута и др. Осадок после отстаивания отфильтровывают, используя обеззоленный фильтр "красная лента" диаметром 12,5 (11,0) см, промывают горячей дистиллированной водой, растворяют в 70 мл 1 н HCl, добавляют 500 - 700 мг лимонной кислоты и аскорбиновую кислоту до обесцвечивания раствора. Раствор доводят дистиллированной водой до 100 мл. В раствор помещают диск из никелевой фольги (рекомендуется применять диски диаметром 34 мм и толщиной 0,6 мм), закрепленный в тефлоновой кассете, которая обеспечивает выделение 210Po на одной стороне диска. Нагревают содержимое стакана на кипящей водяной бане в течение 6 ч, периодически перемешивая. Объем жидкости должен быть равным 100 мл в течение всего времени выделения, поэтому необходимо добавлять к пробе дистиллированную воду по мере ее испарения. Осаждение полония сильно зависит от условий проведения процесса, поэтому все требования должны строго соблюдаться.
Через 6 часов вынимают диск из тефлоновой кассеты, промывают дистиллированной водой, этиловым спиртом и высушивают на воздухе. Записывают время - это время отделения 210Bi от 210Pb. Раствор, оставшийся после электрохимического осаждения изотопов полония и висмута, содержащий изотопы урана и тория (раствор 4), оставляют для дальнейшего определения этих радионуклидов.
Измерение активности 210Po и 210Bi в приготовленном счетном образце и определение их удельной активности в пробе воды выполняют в соответствии с п. 13.5.
После измерения активности диски дезактивируют, для чего их необходимо прокипятить в течение 1 часа с 0,5 н HNO3, промыть проточной водой, обработать мелкой наждачной бумагой, промыть дистиллированной водой и этиловым спиртом или ацетоном. Такая обработка обеспечивает необходимую дезактивацию дисков.
Примечание:
Для дополнительного контроля полноты выделения 210Po и 210Bi в отдельных пробах периодически выборочно в раствор, полученный после первого выделения 210Po и 210Bi, помещают тефлоновую кассету с новым диском и содержимое выдерживают на кипящей водяной бане в течение 1 часа при помешивании. Затем вынимают диск, промывают водой, спиртом, высушивают на воздухе и выполняют измерение ![]() - и
- и ![]() -активности счетного образца.
-активности счетного образца.
В том случае, если активность радионуклидов на втором диске не превышает 5% от активности на первом диске, за результат измерений принимают активность 210Po и 210Bi на первом диске, вычисленную по формулам:
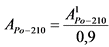 , (2)
, (2)
где: 0,9 - химический выход 210Po, отн. ед.
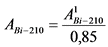 , (3)
, (3)
где: 0,85 - химический выход 210Bi, отн. ед.
В противном случае проводят выделение 210Po и 210Bi на третьем диске с последующим измерением активности радионуклидов.
Активность радионуклидов в счетном образце вычисляют по формулам:
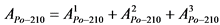 , (4)
, (4)
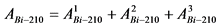 . (5)
. (5)
12.3. Радиохимическое выделение изотопов урана
Природный уран состоит из трех изотопов, однако в значимых количествах в природных водах и других объектах окружающей среды доминируют, как правило, 238U и 234U.
В применяемой в данных Рекомендациях схеме анализа предусмотрено два варианта выделения, очистки и измерения активности изотопов урана.
12.3.1. По первому варианту с использованием радиометра типа УМФ-2000 определяют 238U. Определение 238U производят по дочернему 234Th. Выделение 234Th основано на соосаждении его с оксалатом иттрия. Для этого в раствор, оставшийся после электрохимического осаждения на никелевом диске 210Po и 210Bi (раствор 4), вносят хлористый иттрий (около 60 мг в расчете на Y2O3). Для разрушения лимонной и аскорбиновой кислот раствор упаривают в стакане, закрытом часовым стеклом, до влажных солей, добавляя по 10 - 15 мл смеси концентрированных соляной и азотной кислот в объемном соотношении 1:1. Операцию повторяют 3 - 5 раз.
Влажные соли обрабатывают несколько раз концентрированной HCl для удаления HNO3. Остаток растворяют в 100 мл 1н HCl, нагревают до кипения. Нерастворимый осадок отфильтровывают, используя обеззоленный фильтр "белая лента" диаметром 12,5 см и отбрасывают. К фильтрату добавляют 10 мл насыщенного раствора щавелевой кислоты и аммиаком доводят pH до 1,5. Раствор с осадком оксалата иттрия (тория) охлаждают, оставляют на 1 - 2 часа, затем осадок отфильтровывают через обеззоленный фильтр "синяя лента" диаметром 12,5 см, промывают дистиллированной водой и отбрасывают. Записывают время фильтрования (это время отделения 234Th от 238U). К оставшемуся фильтрату добавляют смесь HCl (30 мл) и HNO3 (30 мл) и упаривают до влажных солей. Операцию повторяют 2 - 3 раза до полного разложения щавелевой кислоты. Влажные соли обрабатывают несколько раз концентрированной HCl для удаления HNO3. Остаток растворяют в 30 мл 2н HCl, разбавляют дистиллированной водой до 80 мл, нагревают, приливают 10 мл 10% раствора (NH4)2CO3, устанавливают pH = 8 - 9 с помощью раствора NH4OH, отфильтровывают выпавший осадок через фильтр "белая лента", промывают 2% раствора углекислого аммония. Осадок отбрасывают. К фильтрату, содержащему растворимый карбонатный комплекс 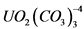 , осторожно приливают смесь HNO3 и HCl (1:1), закрывают часовым стеклом и кипятят до разрушения аммонийных солей (операцию необходимо повторить 2 - 4 раза). Удаление HNO3 проводят обработкой влажных солей концентрированной HCl. Влажные соли растворяют в 30 - 50 мл 2н HCl, разбавляют дистиллированной водой до 100 мл и оставляют на накопление 234Th (время накопления 234Th с учетом его периода полураспада не должно быть менее 24 дней). После накопления 234Th в фильтрат повторно вносят хлористый иттрий, раствор кипятят 20 - 30 минут, после чего проводят вторичное осаждение оксалата иттрия (тория) указанным выше способом. Выделенный осадок оксалата прокаливают при температуре 800 - 900 °C. Окись иттрия растирают до состояния пудры, взвешивают и наносят на подложку для измерения
, осторожно приливают смесь HNO3 и HCl (1:1), закрывают часовым стеклом и кипятят до разрушения аммонийных солей (операцию необходимо повторить 2 - 4 раза). Удаление HNO3 проводят обработкой влажных солей концентрированной HCl. Влажные соли растворяют в 30 - 50 мл 2н HCl, разбавляют дистиллированной водой до 100 мл и оставляют на накопление 234Th (время накопления 234Th с учетом его периода полураспада не должно быть менее 24 дней). После накопления 234Th в фильтрат повторно вносят хлористый иттрий, раствор кипятят 20 - 30 минут, после чего проводят вторичное осаждение оксалата иттрия (тория) указанным выше способом. Выделенный осадок оксалата прокаливают при температуре 800 - 900 °C. Окись иттрия растирают до состояния пудры, взвешивают и наносят на подложку для измерения ![]() -активности препарата.
-активности препарата.
Измерение активности 234Th в приготовленном счетном образце и определение удельной активности 238U в пробе воды выполняют в соответствии с п. 13.6.
12.3.2. По второму варианту определяют 238U и 234U. Метод основан на хроматографическом разделении изотопов урана и тория анионитом ЭДЭ-10П (или другим полифункциональным низкоосновным анионитом конденсационного типа) с последующим ![]() -спектрометрическим определением активности радионуклидов [2].
-спектрометрическим определением активности радионуклидов [2].
В данном случае для определения химического выхода урана и тория на начальном этапе анализа в пробу вводят изотопные индикаторы, в качестве которых используют 232U и 234Th. Активность вносимого 232U не должна превышать ожидаемых в пробе воды активностей 238U и 234U более чем в 5 - 10 раз. Как правило, в пробу вносят 0,2 - 0,3 Бк 232U. Количество вносимого 234Th не должно быть менее 50 Бк/пробу. Для приготовления контрольного раствора изотопного индикатора 234Th используют методику, приведенную в МВИ "Методика выполнения измерений объемной активности изотопов тория (232, 230, 228) в природных водах ![]() -спектрометрическим методом с радиохимической подготовкой" (Приложение 7, пункт 7).
-спектрометрическим методом с радиохимической подготовкой" (Приложение 7, пункт 7).
Раствор, оставшийся после выделения из него 210Po и 210Bi (раствор 4), упаривают до 20 - 30 мл, добавляют 10 мл смеси концентрированных HCl и HNO3 (объемное соотношение 1:1), закрывают часовым стеклом и кипятят до образования влажных солей. Операцию повторяют несколько раз до полного разложения органических веществ (в основном лимонной и аскорбиновой кислот). Влажные соли обрабатывают концентрированной HCl до удаления HNO3, растворяют в 50 мл 8 н HCl и пропускают раствор через колонку диаметром 10 мм с 15 мл анионообменной смолы ЭДЭ-10П со скоростью 1 мл/мин. Затем через колонку пропускают 70 мл 8 н HCl. Объединенные элюаты (раствор 5), содержащие изотопы тория, оставляют для дальнейшего определения этих радионуклидов.
Изотопы урана элюируют с колонки 0,5 н HCl. Объем элюата должен составлять 100 - 150 мл, что обеспечивает полноту десорбции урана. Полученный раствор упаривают до 10 - 20 мл, приливают 10 мл 10% раствора углекислого аммония, устанавливают pH = 8 - 9 с помощью раствора аммиака, нагревают до коагуляции осадка, затем отфильтровывают осадок через фильтр "белая лента", промывают 5 мл 2% раствора углекислого аммония. Далее проводят переосаждение осадка Fe(OH)3 для более полного отделения изотопов урана от мешающих элементов. Для этой цели отфильтрованный и промытый осадок Fe(OH)3 растворяют в 20 мл 0,5 н HCl и повторяют процесс осаждения Fe(OH)3 в присутствии раствора карбоната аммония. К объединенным фильтратам, содержащим растворимый карбонатный комплекс 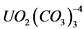 , осторожно приливают смесь HNO3 и HCl (1:1), закрывают часовым стеклом и кипятят до разрушения аммонийных солей (операцию необходимо повторить 2 - 4 раза). Удаление HNO3 проводят обработкой влажных солей концентрированной HCl. К полученному сухому остатку добавляют 10 см3 0,5 н HNO3 и нагревают. К полученному раствору прибавляют 5 см3 1% раствора трилона Б, 1 см3 насыщенного (NH4)2C2O4 и 1 см3 25% NH4Cl. Доводят аммиаком pH до 9, переносят в электролитическую ячейку, стакан обмывают 5 см3 дистиллированной воды. Промывные воды также переносят в электролитическую ячейку.
, осторожно приливают смесь HNO3 и HCl (1:1), закрывают часовым стеклом и кипятят до разрушения аммонийных солей (операцию необходимо повторить 2 - 4 раза). Удаление HNO3 проводят обработкой влажных солей концентрированной HCl. К полученному сухому остатку добавляют 10 см3 0,5 н HNO3 и нагревают. К полученному раствору прибавляют 5 см3 1% раствора трилона Б, 1 см3 насыщенного (NH4)2C2O4 и 1 см3 25% NH4Cl. Доводят аммиаком pH до 9, переносят в электролитическую ячейку, стакан обмывают 5 см3 дистиллированной воды. Промывные воды также переносят в электролитическую ячейку.
Электролиз проводят при силе тока 2 А в течение 0,5 часа. В ходе электролиза контролируют pH раствора, поддерживая его на уровне 9. За минуту до окончания электролиза в раствор добавляют 1 мл раствора аммиака. Разбирают ячейку, вынимают мишень (диск из нержавеющей стали), промывают этиловым спиртом и прокаливают на спиртовке до слегка желтого цвета. Полученный препарат является счетным образцом для измерения активности изотопов урана на ![]() -спектрометре.
-спектрометре.
Примечание:
для дезактивации использованные диски кипятят в течение 1 часа в 0,5 н HNO3, промывают проточной водой, очищают содой и обрабатывают тонкой наждачной бумагой, промывают дистиллированной водой, затем этиловым спиртом или ацетоном. Скорость счета от диска, измеренная на соответствующей аппаратуре, не должна превышать фоновых значений, в противном случае очистку повторяют.
12.3.3. При отсутствии альфа-спектрометра для определения удельной активности 238U и 234U готовят два счетных образца согласно пп. 12.3.1, 12.3.2 Рекомендаций. Толстослойный счетный образец, приготовленный в соответствии с п. 12.3.1 Рекомендаций используют для определения активности 238U по дочернему 234Th. Измерение активности 234Th в приготовленном счетном образце и определение удельной активности 238U в пробе воды выполняют в соответствии с п. 13.6 Рекомендаций.
Тонкослойный счетный образец, приготовленный в соответствии с п. 12.3.2 Рекомендаций, содержит соединения обоих альфа-излучающих изотопов урана: 238U и 234U. Измерение суммарной активности (238U, 234U) и определение их суммарной удельной активности выполняют в соответствии с п. 13.6 Рекомендаций. Удельную активность 234U рассчитывают вычитанием удельной активности 238U из суммарной удельной активности (238U, 234U).
12.4. Радиохимическое выделение изотопов тория
По настоящим Рекомендациям определяют три альфа-излучающих изотопа тория: 232Th, член его радиоактивного семейства - 228Th, а также 230Th, который является членом природного семейства 238U. Для определения удельной активности изотопов тория солянокислый раствор, полученный после отделения изотопов урана (раствор 5), разбавляют дистиллированной водой до кислотности 3 н по HCl. Приготовленный солянокислый раствор пропускают через колонку, заполненную катионитом КУ-2-8 со скоростью 1 мл/мин. При этом на смоле сорбируются изотопы тория, одновременно происходит отделение основных макрокомпонентов. Для очистки от ионов железа и некоторых других катионов смолу промывают 100 мл 3 н HCl с той же скоростью. Десорбцию изотопов тория проводят с помощью раствора оксалата или карбоната аммония, который образует с торием отрицательно заряженный карбонатный комплекс. Для этого через смолу пропускают 100 мл 10% раствора оксалата или карбоната аммония. Первую порцию приливают очень осторожно, т.к. происходит бурное разложение карбоната в соляной кислоте, оставшейся на смоле. Можно предварительно вытеснить фронт кислоты из верхней части колонки небольшой (5 - 8 мл) порцией воды.
Элюат, содержащий изотопы тория, выпаривают до влажных солей, приливают 10 - 15 мл смеси концентрированной соляной и азотной кислот (1:1), закрывают часовым стеклом и продолжают выпаривание. Обработку кислотами повторяют до полного разрушения карбоната аммония. Азотную кислоту удаляют выпариванием влажных солей с концентрированной соляной кислотой (несколько раз). Полученный сухой остаток растворяют при нагревании в 10 см3 1% раствора трилона Б, приливают по 1 см3 25% раствора NH4Cl и насыщенного раствора (NH4)2CO3, устанавливают pH 3 - 4 добавлением по каплям 7 н HNO3. Раствор переносят в электролитическую ячейку, стакан обмывают 8 см3 дистиллированной воды. Промывные воды также переносят в электролитическую ячейку.
Электролиз проводят при силе тока 2 А в течение 0,5 часа. В ходе электролиза контролируют pH раствора, поддерживая его на уровне 3 - 4, добавлением по каплям аммиака или 7 н HNO3. За минуту до окончания электролиза в раствор добавляют 1 мл раствора аммиака. Разбирают ячейку, вынимают мишень (диск из нержавеющей стали), промывают этиловым спиртом и прокаливают на спиртовке до слегка желтого цвета. Полученный препарат является счетным образцом для измерения активности изотопов тория на ![]() -спектрометре.
-спектрометре.
Примечание: при отсутствии ![]() -спектрометра можно провести измерение суммарной активности изотопов тория в приготовленном счетном образце радиометрически, в соответствии с п. 13.7. В этом случае раствор изотопного индикатора 234Th в пробу не вносят. Химический выход тория считают равным 0,75 (эта величина была определена экспериментально при разработке методики с использованием образцового раствора).
-спектрометра можно провести измерение суммарной активности изотопов тория в приготовленном счетном образце радиометрически, в соответствии с п. 13.7. В этом случае раствор изотопного индикатора 234Th в пробу не вносят. Химический выход тория считают равным 0,75 (эта величина была определена экспериментально при разработке методики с использованием образцового раствора).
12.5. Радиохимическое выделение 90Y(90Sr) и 137Cs
В отобранную аликвоту воды после подкисления HCl до pH = 1 вносят растворы носителей - хлористый иттрий (60 мг в расчете на Y2O3), хлористый стронций (100 мг в расчете на SrSO4), хлористый цезий (80 мг в расчете на Cs3Sb2I9) и хлористый лантан (60 мг в расчете на La2O3). При малой минерализации воды в пробу необходимо добавить 50 - 100 мг хлористого кальция. Пробу упаривают до 500 - 600 мл. Из горячего раствора осаждают оксалаты щелочно-земельных элементов добавлением насыщенного раствора щавелевой кислоты (или щавелевокислого аммония) и раствора аммиака до pH = 4. Раствор с осадком оставляют на 2 - 4 часа, отфильтровывают осадок и промывают его дистиллированной водой, содержащей оксалат-ионы.
Осадок прокаливают при температуре 600 - 800 °C в течение часа, растворяют в 2 н HCl и кипятят до полного удаления CO2. После этого осаждают гидроокись иттрия (лантана) безугольным аммиаком и отфильтровывают, используя обеззоленный фильтр "красная лента" диаметром 11,0 см. Записывают время отделения 90Y от 90Sr. Осадок растворяют в 30 - 40 мл 1 н HCl, насыщают мелкорастертым сульфатом калия до появления осадка (отделение иттрия от редкоземельных радионуклидов). Через 0,5 часа осадок отфильтровывают, используя обеззоленный фильтр "синяя лента" диаметром 11,0 см, промывают раствором 1 н HCl, насыщенным сульфатом калия и отбрасывают (процедуру осаждения сульфатов калия и лантана повторяют). Фильтрат разбавляют дистиллированной водой до объема 100 мл, осаждают гидроокиси иттрия безугольным аммиаком при pH = 8 - 9 и отфильтровывают, используя обеззоленный фильтр "красная лента" диаметром 11,0 см. Осадок растворяют в 2 н HCl, разбавляют дистиллированной водой до 100 мл, добавляют 10 мл насыщенного раствора щавелевой кислоты и аммиаком доводят pH до 1,5 для образования осадка оксалата иттрия. Через 20 - 30 минут осадок отфильтровывают, используя обеззоленный фильтр "синяя лента" диаметром 12,5 см, промывают дистиллированной водой с оксалат-ионами и прокаливают в муфеле в течение 1 часа при температуре 800 - 900 °C для переведения оксалата иттрия в Y2O3. Полученный осадок наносят равномерным слоем на предварительно взвешенную подложку, взвешивают осадок вместе с подложкой для определения химического выхода иттрия. Измерение активности 90Y в приготовленном счетном образце и определение удельной активности 90Sr в пробе воды выполняют в соответствии с п. 13.8.
В фильтрате (после осаждения оксалатов щелочноземельных и редкоземельных элементов) определяют 137Cs. Для этого в раствор последовательно вносят 5 - 7 мл 10% азотнокислого никеля и 5 - 7 мл 10% ферроцианида натрия, перемешивают 2 - 3 минуты и оставляют на несколько часов (лучше на ночь) для формирования осадка. Осадок ферроцианида никеля, содержащего 137Cs, отфильтровывают, используя обеззоленный фильтр "синяя/белая лента" диаметром 15,0 см, и прокаливают в муфеле при температуре 400 - 500 °C в течение 2 - 3 часов до полного сгорания фильтра. Фильтрат используют для определения 40K. Прокаленный осадок переносят в стакан, заливают 3 н HCl и нагревают до кипения. Нерастворившиеся частицы отфильтровывают, используя обеззоленный фильтр "белая/желтая лента" диаметром 7,0 см, промывают на фильтре 3 н HCl, общий объем раствора не должен быть более 50 мл. Фильтрат охлаждают, вносят в него 3 мл свежеприготовленного насыщенного раствора NH4I, обесцвечивают раствор добавлением сернистокислого натрия, затем приливают 0,8 мл треххлористой сурьмы SbCl3 и тщательно перемешивают раствор стеклянной палочкой до образования осадка Cs3Sb2I9 красного цвета. Осадок выдерживают 1,5 - 2 часа (можно на ледяной бане).
Отстоявшийся осадок Cs3Sb2I9 фильтруют, используя обеззоленный фильтр "синяя лента" или "желтая лента" диаметром 7,0 см, промывают 3 - 4 раза ледяной уксусной кислотой (до осветления промывных вод), затем этиловым спиртом до удаления запаха уксусной кислоты и высушивают. Высушенный осадок наносят равномерным слоем на предварительно взвешенную подложку, взвешивают осадок вместе с подложкой для определения химического выхода цезия.
Измерение активности 137Cs в приготовленном счетном образце и определение его удельной активности в пробе воды выполняют в соответствии с п. 13.9.
12.6. Приготовление счетных образцов для определения удельной активности 40K
Счетный образец для определения удельной активности 40K готовят путем выпаривания до сухого остатка фильтрата, полученного после выделения 90Y(90Sr) и 137Cs. Сухой остаток прокаливают в муфельной печи в течение 1 часа при температуре 300 - 400 °C. Осадок растирают до состояния "пудры", взвешивают, переносят на подложку целиком или берут аликвоту (в зависимости от веса выделенного осадка), фиксируют этиловым спиртом, высушивают под инфракрасной лампой.
Измерение активности 40K в приготовленном счетном образце и определение его удельной активности в пробе воды выполняют в соответствии с п. 13.10.
13. РЕКОМЕНДАЦИИ ПО ИЗМЕРЕНИЮ АКТИВНОСТИ СЧЕТНЫХ
ОБРАЗЦОВ И ОПРЕДЕЛЕНИЮ УДЕЛЬНОЙ АКТИВНОСТИ РАДИОНУКЛИДОВ
В ПРОБАХ ВОДЫ
13.1. Подготовка к измерениям
Для проведения измерения с заранее заданной погрешностью нужно определить время измерения пробы на основании заданной погрешности, скорости счета от фона и времени его измерения, а также ориентировочной скорости счета от препарата. Выбор времени счета препарата производят по формуле:
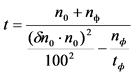 , мин., (6)
, мин., (6)
где:
n0 - скорость счета от препарата за вычетом фона, имп./мин.;
nф - скорость счета от фона, имп./мин.;
![]() - заданная погрешность измерения, %, принимается по данным таблицы 1;
- заданная погрешность измерения, %, принимается по данным таблицы 1;
tф - время измерения фона, мин.;
t - время измерения препарата, мин.
Измерения активности радионуклидов выполняют с применением альфа-бета радиометра, например, УМФ-2000, по схеме:
- проводят подготовку радиометра к работе согласно руководству по эксплуатации прибора;
- устанавливают в барабан радиометра чистую, пустую измерительную кювету и выполняют измерение фона в альфа- и/или бета-канале в течение 60 мин.;
- измерения выполняют в соответствии с техническим описанием и инструкцией по эксплуатации радиометра, например, УМФ-2000;
- время измерения и количество накопленных импульсов (Nф) заносят в рабочий журнал;
- вычисляют фоновую скорость счета импульсов nф, имп./мин., по формуле:
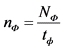 , (7)
, (7)
где:
Nф - количество накопленных импульсов при измерении фона, имп.;
tф - время измерения фона, мин.;
- в случае, если nф превышает фоновое значение в ![]() - и/или
- и/или ![]() -канале, проводят дезактивацию барабана радиометра этиловым спиртом;
-канале, проводят дезактивацию барабана радиометра этиловым спиртом;
- устанавливают в барабан радиометра контрольный источник и выполняют измерение в течение 10 мин.;
- время измерения tки, мин., и количество накопленных импульсов Nки, имп., заносят в рабочий журнал;
- вычисляют скорость счета импульсов от контрольного источника по ![]() - и/или
- и/или ![]() -каналу nки, имп./мин., по формуле:
-каналу nки, имп./мин., по формуле:
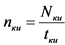 , (8)
, (8)
где:
Nки - количество накопленных импульсов при измерении контрольного источника, имп.;
tки - время измерения контрольного источника, мин.;
- скорость счета импульсов от контрольного источника не должна отличаться от значения, указанного в свидетельстве о поверке, более чем на 5%. В том случае, если это условие не выполняется, выясняют причины, которые могли привести к изменению чувствительности радиометра. В случае необходимости проводят ремонт, наладку прибора. После ремонта проводят поверку прибора.
13.2. Определение удельной суммарной активности ![]() - и
- и ![]() -излучающих радионуклидов
-излучающих радионуклидов
13.2.1. Определяют коэффициент связи радиометра для альфа- и бета-излучения образцов сравнения, изготовленных на основе 226Ra (![]() -канал) и 40K (
-канал) и 40K (![]() -канал) в соответствии с Приложением 5.
-канал) в соответствии с Приложением 5.
13.2.2. Вычисляют скорость счета импульсов от счетного образца по ![]() - и
- и ![]() -каналу nсо, имп./мин., по формуле:
-каналу nсо, имп./мин., по формуле:
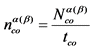 , (9)
, (9)
где:
![]() - количество накопленных импульсов при измерении счетного образца, имп.;
- количество накопленных импульсов при измерении счетного образца, имп.;
tсо - время измерения счетного образца, мин.
13.2.3. Определяют удельную суммарную активность ![]() -излучающих радионуклидов в воде
-излучающих радионуклидов в воде ![]() по формуле:
по формуле:
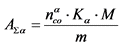 , Бк/кг, (10)
, Бк/кг, (10)
где:
![]() - скорость счета импульсов от образца за вычетом фона по
- скорость счета импульсов от образца за вычетом фона по ![]() -каналу, имп./мин.;
-каналу, имп./мин.;
![]() - коэффициент связи для
- коэффициент связи для ![]() -излучения образца, (Бк/мг)/(имп./мин.);
-излучения образца, (Бк/мг)/(имп./мин.);
M - масса выделенного осадка, мг;
m - масса пробы воды, кг.
13.2.4. Определяют суммарную удельную активность ![]() -излучающих радионуклидов в воде
-излучающих радионуклидов в воде ![]() по формуле:
по формуле:
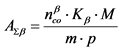 , Бк/кг, (11)
, Бк/кг, (11)
где:
![]() - скорость счета импульсов от образца за вычетом фона по
- скорость счета импульсов от образца за вычетом фона по ![]() -каналу, имп./мин.;
-каналу, имп./мин.;
![]() - коэффициент связи для
- коэффициент связи для ![]() -излучения, Бк/(имп./мин.);
-излучения, Бк/(имп./мин.);
M - масса выделенного осадка, мг; m - масса пробы воды, кг;
p - масса аликвоты выделенного осадка, перенесенная на подложку, мг.
13.3. Определение удельной активности 228Ac(228Ra)
13.3.1. После вычисления скорости счета импульсов от счетного образца La2O3(228Ac) по ![]() -каналу определяют удельную активность 228Ac(228Ra) в пробе воды по формуле:
-каналу определяют удельную активность 228Ac(228Ra) в пробе воды по формуле:
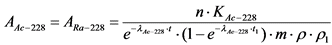 , Бк/кг, (12)
, Бк/кг, (12)
где:
n - скорость счета импульсов от счетного образца за вычетом фона, имп./мин.;
KAc-228 - коэффициент связи для 228Ac, Бк/(имп./мин.), определенный согласно Приложению 5;
![]() - постоянная распада 228Ac;
- постоянная распада 228Ac;
t - время, прошедшее от момента осаждения гидроокиси лантана (отделение 228Ac от 228Ra) до момента начала измерения скорости счета, час.;
t1 - время накопления 228Ac в растворе 228Ra, час. (при условии t1 < 40 часов);
m - масса пробы воды, кг;
![]() - химический выход лантана;
- химический выход лантана;
![]() - химический выход бария.
- химический выход бария.
Величины поправок на распад 228Ac (T1/2 = 6,13 часа) в зависимости от времени приведены в таблице 2.
Таблица 2
Поправка на распад 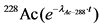 в зависимости от времени
в зависимости от времени
Время распада 228Ac, часы (t)
![]()
1
0,90
1,5
0,84
2
0,79
2,5
0,76
3
0,71
3,5
0,67
4
0,64
4,5
0,60
5
0,57
6
0,50
13.4. Определение удельной активности 226Ra и 224Ra
Изотопы 226Ra и 224Ra имеют сравнительно большие радиоактивные цепочки распада, в результате чего после совместного выделения этих изотопов в полученных препаратах сульфата бария (радия) происходит изменение активности, которое должно обязательно учитываться для корректного определения активности изотопов радия. ![]() -активность 224Ra в выделенном образце возрастает за счет накопления в нем короткоживущих продуктов распада радионуклида (220Rn и 216Po). Максимум активности, в 3,2 раза превышающий исходную величину, достигается через 15 часов. Затем происходит ее спад с T1/2 = 3,6 дня (Таблица 3).
-активность 224Ra в выделенном образце возрастает за счет накопления в нем короткоживущих продуктов распада радионуклида (220Rn и 216Po). Максимум активности, в 3,2 раза превышающий исходную величину, достигается через 15 часов. Затем происходит ее спад с T1/2 = 3,6 дня (Таблица 3).
Таблица 3
Поправка на распад 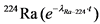 в зависимости от времени
в зависимости от времени
Время распада 224Ra, сутки (t)
![]()
1
0,83
2
0,68
2,5
0,62
3
0,57
3,75
0,49
4
0,47
4,75
0,41
5
0,39
6
0,3
11
0,12
Нарастание ![]() -активности 226Ra в течение первого месяца определяется накоплением 222Rn и его короткоживущих продуктов распада, которые сравнительно быстро приходят с ним в равновесие. Активность 210Po вследствие большого периода полураспада его предшественника 210Pb (T1/2 = 22,3 года) возрастает в первые три месяца медленно и, т.к. она на три порядка меньше общей
-активности 226Ra в течение первого месяца определяется накоплением 222Rn и его короткоживущих продуктов распада, которые сравнительно быстро приходят с ним в равновесие. Активность 210Po вследствие большого периода полураспада его предшественника 210Pb (T1/2 = 22,3 года) возрастает в первые три месяца медленно и, т.к. она на три порядка меньше общей ![]() -активности, то ее можно не учитывать. 210Po приходит в равновесие с 226Ra более чем через 5 лет [3].
-активности, то ее можно не учитывать. 210Po приходит в равновесие с 226Ra более чем через 5 лет [3].
В течение первого месяца к испускаемой 226Ra ![]() -частице добавляются еще три
-частице добавляются еще три ![]() -частицы, испускаемые 222Rn, 218Po и 214Po. Вследствие этого в этот период изменение
-частицы, испускаемые 222Rn, 218Po и 214Po. Вследствие этого в этот период изменение ![]() -активности 226Ra можно описать формулой:
-активности 226Ra можно описать формулой:
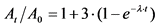 , (13)
, (13)
где:
At - активность 226Ra в момент времени t после отделения его от продуктов распада, Бк/кг;
A0 - активность 226Ra в момент его выделения, когда t = 0, Бк/кг;
t - время от момента отделения 226Ra от продуктов его распада до измерения активности счетного образца, сутки;
![]() - постоянная распада 222Rn.
- постоянная распада 222Rn.
Соответствующие величины, характеризующие изменение ![]() -активности 226Ra в течение первого месяца после выделения радионуклида, представлены в таблице 4.
-активности 226Ra в течение первого месяца после выделения радионуклида, представлены в таблице 4.
Таблица 4
Изменение ![]() -активности 226Ra в первый месяц
-активности 226Ra в первый месяц
после выделения радионуклида (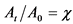 )
)
t, сутки
![]()
t, сутки
![]()
1
1,5
14
3,76
2
1,92
15
3,81
3
2,26
17
3,86
4
2,45
18
3,89
5
2,80
20
3,92
6
2,98
21
3,93
7
3,15
22
3,95
8
3,28
28
3,98
10
3,52
30
3,99
11
3,60
36
4,00
Для определения активности 226Ra счетный образец достаточно измерить через 4 часа, и через 1, 2 и 3 суток после отделения 222Rn и сравнить полученные величины с результатами, рассчитанными по формуле (13).
Совпадение (в пределах погрешности измерения) величин свидетельствует о наличии в образце преимущественно 226Ra и практическом отсутствии 224Ra, что позволяет определить активность 226Ra в период от 4 часов до 2 - 3 суток после отделения 226Ra от 222Rn. В этом случае вычисляют скорость счета импульсов от счетного образца (BaSO4) по ![]() -каналу через 4 часа после прокаливания, а также через 1, 2 и 3 суток. Для скорости счета импульсов, полученной через 1, 2 и 3 суток после прокаливания (т.е. после отделения 226Ra от 222Rn), вносят соответствующие поправки из таблицы (4), берут среднюю из всех временных точек величину и определяют удельную активность 226Ra по формуле:
-каналу через 4 часа после прокаливания, а также через 1, 2 и 3 суток. Для скорости счета импульсов, полученной через 1, 2 и 3 суток после прокаливания (т.е. после отделения 226Ra от 222Rn), вносят соответствующие поправки из таблицы (4), берут среднюю из всех временных точек величину и определяют удельную активность 226Ra по формуле:
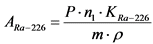 , Бк/кг, (14)
, Бк/кг, (14)
где: P - масса счетного образца, мг;
n1 - средняя скорость счета импульсов от счетного образца через 4 часа, а также (с учетом поправок из таблицы) через 1, 2 и 3 суток после прокаливания BaSO4(226Ra), имп./мин.;
KRa-226 - коэффициент связи для 226Ra, (Бк/мг)/(имп./мин.), определенный согласно Приложению 5;
m - масса пробы воды, кг;
![]() - химический выход бария.
- химический выход бария.
Если величины, полученные при измерении активности счетного образца через 1, 2 и 3 суток после отделения 226Ra от 222Rn, не совпадают с рассчитанными по формуле (13) величинами, то это означает, что в пробе помимо 226Ra, в соизмеримых с ним количествах, присутствует 224Ra. В этом случае для идентификации 226Ra и 224Ra необходимо дополнительное измерение альфа-активности счетного образца через промежуток времени (t), достаточный для распада находящегося в препарате 224Ra. Поскольку период полураспада 224Ra равен 3,64 суток, дополнительное измерение альфа-активности можно проводить, начиная с 11 суток после первого измерения. К этому времени 224Ra практически распадается, а нарастание активности 226Ra учитывают, используя формулу (13).
Удельную активность 226Ra и 224Ra вычисляют соответственно по формулам:
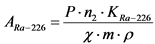 , Бк/кг (15)
, Бк/кг (15)
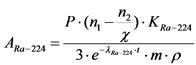 , Бк/кг, (16)
, Бк/кг, (16)
где:
P - масса счетного образца, мг;
n2 - скорость счета импульсов от счетного образца (за вычетом скорости счета фона) через t суток после первого измерения, имп./мин.;
ARa-226 и KRa-224 - коэффициенты связи для 226Ra и 224Ra, (Бк/мг)/(имп./мин.), определенные согласно Приложению 5;
![]() - коэффициент, учитывающий регистрацию радиометром альфа-частиц, испускаемых дочерними продуктами распада 226Ra через t суток после первого измерения (см. таблицу 4);
- коэффициент, учитывающий регистрацию радиометром альфа-частиц, испускаемых дочерними продуктами распада 226Ra через t суток после первого измерения (см. таблицу 4);
m - масса пробы воды, кг;
![]() - химический выход бария;
- химический выход бария;
n1 - скорость счета импульсов от счетного образца (за вычетом скорости счета фона) через 4 часа после прокаливания BaSO4 (226Ra), имп./мин.;
3 - коэффициент, учитывающий регистрацию радиометром ![]() -частиц, испускаемых дочерними продуктами распада 224Ra;
-частиц, испускаемых дочерними продуктами распада 224Ra;
![]() - постоянная распада 224Ra;
- постоянная распада 224Ra;
t - время, прошедшее от момента отделения 224Ra от 228Th (осаждение Ba(Ra)SO4) до момента первого измерения скорости счета счетного образца, сутки.
13.5. Определение удельной активности 210Po, 210Bi(210Pb)
После электролитического осаждения 210Po и 210Bi на никелевый диск определяют скорость счета полученного образца по ![]() - и
- и ![]() -каналу. Измерение активности препарата выполняют не ранее чем через 10 - 12 часов после электрохимического выделения радионуклидов, поскольку кроме 210Po и 210Bi препарат может содержать короткоживущие
-каналу. Измерение активности препарата выполняют не ранее чем через 10 - 12 часов после электрохимического выделения радионуклидов, поскольку кроме 210Po и 210Bi препарат может содержать короткоживущие ![]() -активные (218Po, 214Po, 216Po, 212Po, 215Po, 211Po, 211Bi) и
-активные (218Po, 214Po, 216Po, 212Po, 215Po, 211Po, 211Bi) и ![]() -активные (214Bi, 212Bi) радионуклиды. Выдержка препарата перед его измерением в течение 10 - 12 часов приводит к распаду этих короткоживущих радионуклидов, т.е. устраняет их мешающее влияние.
-активные (214Bi, 212Bi) радионуклиды. Выдержка препарата перед его измерением в течение 10 - 12 часов приводит к распаду этих короткоживущих радионуклидов, т.е. устраняет их мешающее влияние.
Удельную активность 210Po в пробе воды определяют по формуле (17).
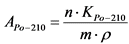 , Бк/кг, (17)
, Бк/кг, (17)
где:
n - скорость счета импульсов от счетного образца за вычетом фона по ![]() -каналу, имп./мин.;
-каналу, имп./мин.;
KPo-210 - коэффициент связи для 210Po, Бк/(имп./мин.), полученный при использовании контрольного образца сравнения;
m - масса пробы воды, кг;
![]() - химический выход 210Po.
- химический выход 210Po.
Удельную активность 210Bi в воде, которую принимают равной удельной активности 210Pb, определяют по формуле:
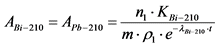 , Бк/кг, (18)
, Бк/кг, (18)
где:
n1 - скорость счета импульсов от счетного образца за вычетом фона по ![]() -каналу, имп./мин.;
-каналу, имп./мин.;
KBi-210 - коэффициент связи для 210Bi, Бк/(имп./мин.), полученный при использовании контрольного образца сравнения;
m - масса пробы воды, кг;
![]() - химический выход 210Bi;
- химический выход 210Bi;
t - время от электрохимического выделения 210Bi до измерения его активности, часы;
![]() - постоянная распада 210Bi.
- постоянная распада 210Bi.
Примечание:
1. Химические выходы 210Po и 210Bi были определены экспериментально при разработке методики с использованием соответствующих стандартных растворов:
![]() = 0,9;
= 0,9; ![]() = 0,85.
= 0,85.
2. Поправки на распад 210Bi (T1/2 = 120 час.) представлены в таблице 5.
Таблица 5
Поправка на распад 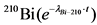 в зависимости от времени
в зависимости от времени
Время распада 210Bi, часы (t)
![]()
12
0,93
16
0,91
20
0,89
24
0,87
28
0,85
32
0,83
40
0,80
13.6. После выделения накопившегося из урана-238 234Th и измерения скорости счета образца на малофоновом альфа-бета радиометре по бета-каналу удельную активность 238U в воде определяют по формуле:
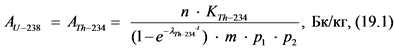
где:
![]() - скорость счета импульсов от счетного образца за вычетом фона по бета-каналу, имп/мин;
- скорость счета импульсов от счетного образца за вычетом фона по бета-каналу, имп/мин;
![]() - коэффициент связи для 234Th, Бк/(имп/мин), определенный согласно приложению 5 к настоящим Рекомендациям;
- коэффициент связи для 234Th, Бк/(имп/мин), определенный согласно приложению 5 к настоящим Рекомендациям;
![]() - постоянная распада 234Th;
- постоянная распада 234Th;
![]() - время накопления 234Th в растворе 238U, сутки;
- время накопления 234Th в растворе 238U, сутки;
![]() - масса пробы воды, кг;
- масса пробы воды, кг;
![]() - химический выход иттрия (234Th);
- химический выход иттрия (234Th);
![]() - химический выход 238U.
- химический выход 238U.
Примечание:
Химический выход 238U был определен экспериментально при разработке методики с использованием стандартного раствора 232U: ![]() = 0,65.
= 0,65.
После электролитического осаждения 238U и 234U на диск из нержавеющей стали и измерения скорости счета образца на малофоновом альфа-бета радиометре по альфа-каналу суммарную удельную активность альфа-излучающих изотопов урана в воде определяют по формуле:
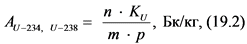
где:
![]() - скорость счета импульсов от счетного образца за вычетом фона по альфа-каналу, имп/мин;
- скорость счета импульсов от счетного образца за вычетом фона по альфа-каналу, имп/мин;
![]() - коэффициент связи для (238U, 234U), Бк/(имп/мин), полученный при использовании контрольного образца сравнения (210Po);
- коэффициент связи для (238U, 234U), Бк/(имп/мин), полученный при использовании контрольного образца сравнения (210Po);
![]() - масса пробы воды, кг;
- масса пробы воды, кг;
![]() - химический выход (238U, 234U).
- химический выход (238U, 234U).
Примечание:
Химический выход (238U, 234U) был определен экспериментально при разработке методики с использованием стандартного раствора 232U: ![]() = 0,65.
= 0,65.
Удельную активность 234U в пробе воды определяют по формуле:
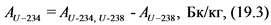
где:
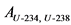 - удельная активность (238U, 234U), Бк/кг, определенная по формуле 19.2;
- удельная активность (238U, 234U), Бк/кг, определенная по формуле 19.2;
![]() - удельная активность 238U, Бк/кг, определенная по формуле 19.1.
- удельная активность 238U, Бк/кг, определенная по формуле 19.1.
13.7. Определение удельной суммарной активности изотопов тория (228Th, 230Th, 232Th)
Вычисляют скорость счета импульсов от счетного образца (в тонком слое) по ![]() -каналу и определяют суммарную удельную активность изотопов тория в пробе воды по формуле:
-каналу и определяют суммарную удельную активность изотопов тория в пробе воды по формуле:
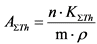 , Бк/кг, (20)
, Бк/кг, (20)
где:
n - скорость счета импульсов от счетного образца за вычетом фона, имп./мин.;
![]() - коэффициент связи для суммы изотопов тория, Бк/(имп./мин.);
- коэффициент связи для суммы изотопов тория, Бк/(имп./мин.);
m - масса пробы, кг;
![]() - химический выход радионуклидов.
- химический выход радионуклидов.
Примечание:
Химический выход суммы ![]() -излучающих изотопов тория определяют, измерив скорость счета образца (234Th) на УМФ-2000 по
-излучающих изотопов тория определяют, измерив скорость счета образца (234Th) на УМФ-2000 по ![]() -каналу.
-каналу.
13.8. Определение удельной активности 90Sr
После вычисления скорости счета импульсов от счетного образца Y2O3(90Y) по ![]() -каналу определяют удельную активность 90Y(90Sr) в пробе воды по формуле:
-каналу определяют удельную активность 90Y(90Sr) в пробе воды по формуле:
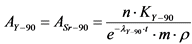 , Бк/кг, (21)
, Бк/кг, (21)
где:
n - скорость счета импульсов от счетного образца за вычетом фона, имп./мин.;
KY-90 - коэффициент связи между активностью 90Y и скоростью счета импульсов от счетного образца, Бк/(имп./мин.), определенный согласно Приложению 5;
![]() - постоянная распада 90Y;
- постоянная распада 90Y;
t - время, прошедшее от момента отделения 90Y от 90Sr до времени измерения активности образца, час.;
m - масса пробы воды, кг;
![]() - химический выход 90Y.
- химический выход 90Y.
Величины поправок на распад 90Y (T1/2 = 64,1 часа) в зависимости от времени приведены в таблице 6.
Таблица 6
Поправка на распад  в зависимости от времени
в зависимости от времени
Время распада 90Y, часы (t)
![]()
Время распада 90Y, часы (t)
![]()
2
0,98
45
0,62
5
0,94
50
0,58
17
0,83
64
0,50
21
0,80
72
0,47
25
0,76
96
0,35
30
0,72
120
0,27
36
0,68
144
0,31
Для проверки радиохимической чистоты выделенного 90Y счетный образец измеряют дополнительно через периоды времени, равные 1 - 2 периодам полураспада радионуклида. Скорость счета, отнесенная к величине поправки на распад в зависимости от времени, прошедшего после отделения 90Y от 90Sr, должна совпадать в пределах ошибки измерения со скоростью счета первого измерения.
13.9. Определение удельной активности 137Cs
После вычисления скорости счета импульсов от счетного образца (Cs3Sb2I9) по ![]() -каналу определяют удельную активность 137Cs в пробе воды по формуле:
-каналу определяют удельную активность 137Cs в пробе воды по формуле:
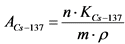 , Бк/кг, (22)
, Бк/кг, (22)
где:
n - скорость счета импульсов от счетного образца за вычетом фона, имп./мин.;
KCs-137 - коэффициент связи между активностью 137Cs и скоростью счета импульсов от счетного образца, Бк/(имп./мин.), определенный согласно Приложению 5;
m - масса пробы воды, кг;
![]() - химический выход 137Cs.
- химический выход 137Cs.
13.10. Определение удельной активности 40K
Счетный образец, приготовленный согласно главе 11 Рекомендаций, помещают в держатель образцов альфа-бета радиометра на посадочное место для измерения проб на толстых подложках, закрывают сверху алюминиевой фольгой толщиной 81 мг/см2 и проводят измерение. Поскольку максимальная энергия бета- излучения природных радионуклидов рядов 238U и 232Th значительно ниже, чем у 40K, за поглощающим экраном из алюминия толщиной 81 мг/см2, соответствующей слою половинного ослабления бета- излучения 40K, регистрируется только бета-излучение 40K, ослабленное в 2 раза.
После вычисления скорости счета импульсов от счетного образца, закрытого алюминиевой фольгой толщиной 81 мг/см2, определяют удельную активность 40K в пробе воды по формуле:
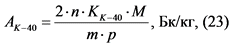
где: n - скорость счета импульсов от счетного образца, закрытого алюминиевой фольгой (толщиной 81 мг/см2) за вычетом фона, имп/мин;
KK-40 - коэффициент связи между активностью 40K и скоростью счета импульсов от счетного образца, Бк/ (имп/мин), определенный согласно приложению 5 к настоящим Рекомендациям;
M - масса выделенного осадка, мг;
m - масса пробы воды, кг;
p - масса аликвоты выделенного осадка, перенесенной на подложку, мг.
В случае, если проба воды, отобрана на территории, предположительно загрязненной техногенным радионуклидом 90Sr(90Y) в количествах, сравнимых с содержанием в воде 40K, определять удельную активность 40K, используя счетный образец, подготовленный для суммарных показателей, нельзя, поскольку максимальная энергия бета- излучения 90Sr(90Y) близка к максимальной энергии бета-излучения 40K. В данном случае, счетный образец для определения удельной активности 40K (по формуле 23) подготавливают согласно пп. 12.5, 12.6 Рекомендаций. При способе подготовки пробы перед измерением активности 40K приведенном в пп. 12.5, 12.6 Рекомендаций, 90Sr(90Y) радиохимически удаляют из пробы на предыдущем этапе анализа. При таком способе подготовки счетного образца бета-излучение 90Sr(90Y), мешающее определению активности 40K, отсутствует.
13.11. Обработка результатов измерений
13.11.1. При полном радионуклидном анализе следует выполнять оценку соответствия удельной суммарной ![]() -активности и суммы активностей радионуклидов (МУ 2.6.1.1981-05) по критерию:
-активности и суммы активностей радионуклидов (МУ 2.6.1.1981-05) по критерию:
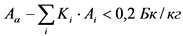 , (24)
, (24)
где:
![]() - удельная суммарная
- удельная суммарная ![]() -активность, Бк/кг;
-активность, Бк/кг;
Ki - коэффициенты, характеризующие несоответствие энергетических спектров стандартов сравнения и реальной пробы (табл. 7);
0,2 - эмпирический коэффициент, учитывающий присутствие в пробе воды других ![]() -излучающих радионуклидов на уровне не более 5% от значения УВi, определение которых в процессе анализа не проводили (например, 234U, 232Th, 230Th, 228Th).
-излучающих радионуклидов на уровне не более 5% от значения УВi, определение которых в процессе анализа не проводили (например, 234U, 232Th, 230Th, 228Th).
Таблица 7
Значения коэффициента Ki при использовании стандарта
сравнения с 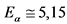 МэВ и нижним уровнем дискриминации
МэВ и нижним уровнем дискриминации
![]() -радиометра
-радиометра ![]() 3 МэВ
3 МэВ
Альфа-излучающий радионуклид
Энергия ![]() -излучения, кэВ
-излучения, кэВ
Значение коэффициента Ki
232Th
4010
0,60
238U
4195
0,65
230Th
4685
0,85
234U, 226Rа
4770; 4780
0,90
239 + 240Pu; 210Po
5155 + 5168; 5305
1,00
228Th; 241Am; 238Pu
5420; 5486; 5500
1,10
224Ra
5680
1,15
13.11.2. Если условие (24) выполнено, то считается, что все основные ![]() -излучающие радионуклиды, содержащиеся в пробе, определены и дальнейшие исследования воды не требуются.
-излучающие радионуклиды, содержащиеся в пробе, определены и дальнейшие исследования воды не требуются.
13.11.3. Результаты определения удельной активности (как и удельной суммарной активности) радионуклидов в пробе воды представляют в форме:
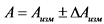 , Бк/кг, (25)
, Бк/кг, (25)
где:
Aизм - результат измерения;
![]() - абсолютная погрешность измерения, численное значение которой рассчитывают по формуле:
- абсолютная погрешность измерения, численное значение которой рассчитывают по формуле:
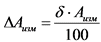 , Бк/кг (26)
, Бк/кг (26)
Значение ![]() принимают либо по таблице 1 (стр. 11), либо рассчитывают, учитывая погрешности, возникающие на всех стадиях радиохимического анализа. Систематические погрешности складываются из:
принимают либо по таблице 1 (стр. 11), либо рассчитывают, учитывая погрешности, возникающие на всех стадиях радиохимического анализа. Систематические погрешности складываются из:
- относительной погрешности определения химического выхода ![]() . В случае выделения изотопа без носителя включается относительная погрешность выхода изотопа, найденная в предварительных экспериментах;
. В случае выделения изотопа без носителя включается относительная погрешность выхода изотопа, найденная в предварительных экспериментах;
- относительной погрешности градуировки прибора, ![]() , суммирующейся из паспортной погрешности определения активности эталона, погрешности при отборе аликвотной части раствора и погрешности измерения активности эталонного раствора.
, суммирующейся из паспортной погрешности определения активности эталона, погрешности при отборе аликвотной части раствора и погрешности измерения активности эталонного раствора.
Среднеквадратическую погрешность измерения определяют по формуле:
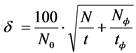 , %, (27)
, %, (27)
где:
N0 - скорость счета от препарата за вычетом фона, имп./мин.;
N - скорость счета от препарата с фоном, имп./мин.;
t - время измерения препарата с фоном, мин.;
Nф - скорость счета от фона, имп./мин.;
tф - время измерения фона, мин.
Среднеквадратическая погрешность зависит от времени измерения и не должна превышать 30%.
Общую погрешность, ![]() , анализа рассчитывают по формуле:
, анализа рассчитывают по формуле:
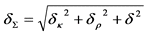 (28)
(28)
14. КОНТРОЛЬ КАЧЕСТВА РЕЗУЛЬТАТОВ ИЗМЕРЕНИЙ ПРИ РЕАЛИЗАЦИИ
МЕТОДИКИ В ЛАБОРАТОРИИ
14.1. Контроль качества результатов измерений при реализации методики в лаборатории предусматривает:
- контроль исполнителем процедуры выполнения измерений (на основе оценки погрешности при реализации отдельно взятой контрольной процедуры);
- контроль стабильности результатов измерения (на основе оценки погрешности при реализации всего анализа в соответствии со схемой, приведенной в Приложении 6).
14.2. Алгоритм контроля процедуры выполнения измерений с использованием образцов для контроля (аттестованных смесей).
14.2.1. Контроль исполнителем процедуры выполнения измерений проводят путем сравнения результата отдельно взятой контрольной процедуры (КК) с нормативом контроля (К).
14.2.2. Результат контрольной процедуры (КК) рассчитывают по формуле:
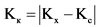 , (29)
, (29)
где:
КХ - результат контрольного измерения активности радионуклида в образце для контроля;
Кс - аттестованное значение образца для контроля.
Качество контрольной процедуры признают удовлетворительным при выполнении условия: К < ![]() , значения
, значения ![]() принимают по таблице 1 в соответствии с аттестованным значением образца для контроля.
принимают по таблице 1 в соответствии с аттестованным значением образца для контроля.
При невыполнении условия (29) эксперимент повторяют. При повторном невыполнении условия (29) выясняют причины, приводящие к неудовлетворительным результатам.
14.2.3. Периодичность контроля исполнителем процедуры выполнения измерений, а также реализуемые процедуры контроля стабильности результатов выполняемых измерений регламентируют в Руководстве по качеству лаборатории.
Приложение 1
к МР 2.6.1.0064-12
(справочное)
РЯДЫ РАДИОАКТИВНОГО РАСПАДА
Радиоактивный ряд урана-238
Радиоактивный ряд тория-232
Приложение 2
к МР 2.6.1.0064-12
(справочное)
ИНСТРУКЦИЯ ПО ОТБОРУ ПРОБ ВОДЫ ИЗ РАЗНЫХ ИСТОЧНИКОВ
1. Отбор проб воды для анализа радиологических показателей следует производить с учетом общих требований ГОСТ Р 51592-2000.
2. Для отбора проб воды используют емкости из полимерных материалов, которые непосредственно перед отбором пробы необходимо не менее трех раз ополоскать водой из обследуемого источника.
3. Масса отбираемой пробы воды должна составлять 1 кг при анализе суммарных показателей, 10 кг - при анализе радионуклидного состава и удельной активности природных радионуклидов в воде и не менее 4 кг при анализе удельной активности 137Cs и 90Sr в воде (4 кг на аварийно загрязненных территориях и 20 кг для фоновых значений).
4. При отборе проб из действующей артезианской скважины или водопровода воду сливают не менее 5 мин., во всех остальных случаях (вновь вводимых, после ремонта и др.) - в течение времени, необходимого для установления стабильных характеристик воды. Пробы воды из колодцев и открытых водоемов отбирают традиционным для данного населенного пункта способом. Отобранную пробу выдерживают в емкости в течение времени, необходимого для осаждения мелкодисперсных частиц песка, почвы или ила, которые могут попасть в пробу.
5. Отобранную пробу подкисляют соляной кислотой до pH = 1 для исключения процесса сорбции микроколичеств радионуклидов.
6. Каждую отобранную пробу опечатывают, снабжают этикеткой, наклеенной на емкость с пробой, и актом отбора. В акте отбора проб должна содержаться вся информация, необходимая для идентификации источника водоснабжения, рекомендуемая информация приведена в Приложении 3 к настоящим Рекомендациям.
7. Пробы должны своевременно пройти процедуру пробоподготовки. Срок хранения подкисленных соляной кислотой проб, помещенных в емкости из полимерного материала, не должен превышать 14 дней.
Приложение 3
к МР 2.6.1.0064-12
(рекомендуемое)
ИНФОРМАЦИЯ ДЛЯ ВНЕСЕНИЯ В АКТ ОТБОРА ПРОБ ВОДЫ
На каждую пробу воды должен быть составлен акт отбора. При одновременном отборе нескольких проб допускается сведения о них приводить в одном акте.
В акте отбора проб воды должна быть отражена следующая информация:
1. Наименование и юридический адрес заказчика;
2. Наименование и адрес источника водоснабжения (артезианская скважина, водопровод, партия бутилированной воды и т.д.) с указанием его основных характеристик (например, при отборе проб бутилированной воды следует указать N партии и дату розлива, вид тары и ее объем, при отборе проб из артезианской скважины - N, глубину и наименование организации владельца скважины);
3. Дату и время отбора пробы;
4. Место отбора пробы;
5. Объем каждой пробы;
6. ФИО и подпись лиц, проводивших отбор проб, с указанием места работы и занимаемой должности;
7. Основание для отбора проб (по заявке заказчика, по предписанию органов Роспотребнадзора и т.п.);
8. Цель отбора проб воды (определение удельной суммарной ![]() - и
- и ![]() -активности, определение удельных активностей природных радионуклидов и т.п.);
-активности, определение удельных активностей природных радионуклидов и т.п.);
9. Характеристика условий отбора проб.
Пробы должны быть опечатаны после их отбора, а также после их консервации и/или концентрирования перед отправкой на исследование.
Приложение 4
к МР 2.6.1.0064-12
(справочное)
ПРИГОТОВЛЕНИЕ
РАБОЧИХ РАСТВОРОВ И РАСТВОРОВ НОСИТЕЛЕЙ. ПОДГОТОВКА
ИОНООБМЕННЫХ СМОЛ К АНАЛИЗУ, РЕГЕНЕРАЦИЯ И ХРАНЕНИЕ
Приготовление рабочих растворов
1. Кислоты:
HCl - 0,5 н; 0,7 н; 1 н; 2 н; 3 н; 8 н.
HNO3 - 0,5 н; 7 н.
Для приготовления рабочего раствора кислоты концентрированную кислоту разбавляют дистиллированной водой до нужной концентрации. Концентрацию определяют, измерив соответствующим ареометром ее удельный вес. Затем, используя формулу (30), вычисляют нормальность кислоты, для чего предварительно определяют ее процентное содержание по справочной литературе, например, [4] и ее эквивалентный вес (таблица 8).
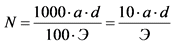 , (30)
, (30)
где:
N - нормальность кислоты;
a - процентное содержание кислоты;
d - удельный вес кислоты;
Э - эквивалентный вес кислоты.
Таблица 8
Эквивалентный вес кислот
Кислота
Эквивалентный вес
Соляная (HCl)
36,47
Азотная (HNO3)
63,02
Серная (H2SO4)
49,04
Определив нормальность кислоты, рассчитывают количество ее, необходимое для приготовления нужного раствора.
Пример: Необходимо приготовить 500 мл 0,5 н HNO3.
Сначала определяют удельный вес исходной кислоты (d = 1,40), затем по справочнику находят, что в азотной кислоте, удельный вес которой 1,40, процентное содержание HNO3 - 67%. Рассчитывают нормальность имеющейся кислоты:
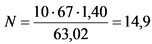
Определяют, во сколько раз имеющаяся кислота концентрированнее приготовляемой: 14,9 : 0,5 = 29,8.
На основании этого устанавливают, что для приготовления 1,0 л 0,5 н HNO3 необходимо разбавить дистиллированной водой до 1,0 л 33,6 мл (1000 мл : 29,8 мл = 33,6 мл) азотной кислоты плотностью d = 1,40.
2. Натрий углекислый, 5% раствор: 50 г соли растворяют при нагревании в 950 см3 дистиллированной воды. Срок хранения 1 год.
3. Лимонная кислота, 1% раствор: 1 г кислоты растворяют в 99 мл дистиллированной воды. Срок хранения 1 год.
4. Аммоний хлористый, 25% раствор: 25 г соли NH4Cl растворяют в 75 см3 дистиллированной воды. Срок хранения не ограничен.
5. Аммоний щавелевокислый, насыщенный раствор: 12 г соли (NH4)2C2O4·H2O растворяют в 100 см3 дистиллированной воды. Срок хранения не ограничен.
6. Щавелевая кислота, насыщенный раствор: 10 г H2C2O4·2H2O растворяют в 100 см3 дистиллированной воды. Срок хранения не ограничен.
7. Серная кислота, 10% раствор: приготовляют по объему растворением 100 мл концентрированной H2SO4 в 900 мл дистиллированной воды. Срок хранения не ограничен.
8. Аммоний углекислый, 15% раствор: 15 г (NH4)2CO3·H2O растворяют в 85 см3 дистиллированной воды. Срок хранения не ограничен.
9. Аммоний углекислый, 10% раствор: 10 г (NH4)2CO3·H2O растворяют в 90 см3 дистиллированной воды. Срок хранения не ограничен.
10. Аммоний углекислый, 2% раствор: 2 г (NH4)2CO3·H2O растворяют в 98 см3 дистиллированной воды. Срок хранения не ограничен.
11. Никель азотнокислый, 10% раствор: 50 г соли растворяют в 450 см3 дистиллированной воды. Срок хранения не ограничен.
12. Натрий железистосинеродистый, 10% раствор: 50 г соли растворяют в 450 см3 дистиллированной воды. Срок хранения не ограничен.
13. Треххлористая сурьма, раствор: 6,9 г SbCl3 растворяют в 100 мл 4 н HCl (или 4,4 г Sb2O3 растворяют в 100 мл 4 н HCl). Срок хранения 1 год.
14. Йодистый аммоний, насыщенный раствор: 170 г соли растворяют в 100 см3 дистиллированной воды. Срок хранения 1 месяц.
15. Оксалатно-аммиачно-хлоридный буферный раствор: 1,26 г оксалата аммония и 53,5 г хлорида аммония растворяют в 1 л дистиллированной воды. Срок хранения не ограничен.
16. Аммиак водный безугольный, раствор: получают перегонкой NH4OH в аппарате, собранном из перегонной колбы, холодильника (ловушки) и приемной колбы. Для связывания CO2 в перегонную колбу добавляют (OH)2 из расчета 5 - 10 г/л. Срок хранения 1 месяц.
Приготовление растворов носителей
Использование носителей значительно упрощает радиохимический анализ, позволяя применять для выделения радионуклидов реакции осаждения труднорастворимых солей и контролировать полноту выделения изотопов. В процессе подготовки пробы к анализу и операций по отделению изотопа от примесей происходят его неконтролируемые потери. Количество радионуклида, прошедшее через весь анализ и измеренное по его радиоактивности, составляет лишь долю от первоначального. Для определения значения этой доли в пробу на самом первом этапе ее обработки добавляют известное количество носителя. Отношение количества носителя, измеренного на выходе, к количеству носителя, добавленного в пробу, дает величину химического выхода изотопа.
Носителями радиоактивных изотопов обычно служат стабильные изотопы определяемых радионуклидов, добавляемые в пробу в виде растворов тех или иных солей, как правило, соляно- или азотнокислых. Количество вводимого носителя зависит от принятых условий измерения радиоактивности препарата. При определении ![]() -излучателей следует стремиться к уменьшению количества носителя, т.к. это приводит к получению препарата с более высокой удельной активностью и к соответствующему уменьшению коэффициента связи между скоростью счета и активностью препарата. Обычно для
-излучателей следует стремиться к уменьшению количества носителя, т.к. это приводит к получению препарата с более высокой удельной активностью и к соответствующему уменьшению коэффициента связи между скоростью счета и активностью препарата. Обычно для ![]() -излучателей количество носителя выбирают равным 50 - 60 мг в пересчете на весовую форму, в виде которой носитель выделяют из пробы и взвешивают (при измерении
-излучателей количество носителя выбирают равным 50 - 60 мг в пересчете на весовую форму, в виде которой носитель выделяют из пробы и взвешивают (при измерении ![]() -активности на подложках площадью 2,5 см2). В этих условиях для определения
-активности на подложках площадью 2,5 см2). В этих условиях для определения ![]() -излучателей в толстом слое количество вводимого в пробу носителя в пересчете на форму взвешивания составляет 80 - 100 мг.
-излучателей в толстом слое количество вводимого в пробу носителя в пересчете на форму взвешивания составляет 80 - 100 мг.
Если объем взятой на анализ пробы велик, необходимо помимо носителей радионуклидов добавлять коллекторный носитель, от которого носитель определяемого изотопа легко отделить в последующих химических операциях. Чаще всего в качестве коллекторного носителя используют соли железа.
Хотя наиболее часто для определения химического выхода изотопа применяют весовой метод, т.к. проще всего измерить радиоактивность твердых препаратов, контроль за потерями радиоактивных изотопов в ходе анализа определяют не только выходом носителя. В некоторых случаях для определения химического выхода можно воспользоваться другим радиоактивным изотопом этого же элемента - метод радиоактивных индикаторов. Когда у элемента нет изотопов, различающихся типом излучения, при использовании метода радиоактивных индикаторов для определения химического выхода необходима сложная аппаратура, например спектрометрическая. При использовании ![]() -спектрометрии препараты для измерения должны быть тонкослойными.
-спектрометрии препараты для измерения должны быть тонкослойными.
Метод радиоактивных индикаторов находит широкое применение при определении ![]() -спектрометрическим методом изотопов урана (234U, 238U), тория (228Th, 230Th, 232Th), а также 210Po. При этом в качестве изотопных индикаторов полноты выделения используются 234Th (
-спектрометрическим методом изотопов урана (234U, 238U), тория (228Th, 230Th, 232Th), а также 210Po. При этом в качестве изотопных индикаторов полноты выделения используются 234Th (![]() -излучатель), 232U и 209Po, энергии
-излучатель), 232U и 209Po, энергии ![]() -частиц которых отличаются от энергий
-частиц которых отличаются от энергий ![]() -частиц определяемых изотопов.
-частиц определяемых изотопов.
Все растворы носителей должны быть либо солянокислыми, либо азотнокислыми и не должны содержать других анионов, например, ![]() . Количество приготовленного носителя зависит от числа проводимых в лаборатории анализов.
. Количество приготовленного носителя зависит от числа проводимых в лаборатории анализов.
Все мерные колбы с растворами носителей должны быть закрыты резиновыми пробками соответствующих размеров и снабжены этикетками с указанием титра носителя и даты его приготовления. Титры носителей необходимо проверять два раза в год.
Для приготовления титрованного раствора любого носителя необходимо:
- рассчитать соотношение молекулярных масс соединения носителя, из которого приготовлен раствор, и соединения носителя, которое применяется в качестве счетного образца при измерении активности радионуклида;
- проводить определение титра носителя в условиях, аналогичных тем, которые используются на последней стадии радиохимического анализа при выделении данного носителя (радионуклида).
Ниже приведены методики приготовления носителей для определения весовым методом химического выхода радионуклидов при определении их удельной активности в воде.
1. Приготовление титрованного раствора бария
Исходная соль для приготовления раствора - BaCl2·2H2O. Соединение, в виде которого выделяется носитель на последней стадии анализа, - BaSO4. Объем приготовляемого раствора носителя - 100 мл. Заданный титр BaSO4 - 100 мг/мл, т.е. в 100 мл раствора должно содержаться 100 мг x 100 мл = 10 г BaSO4.
Молекулярная масса BaCl2·2H2O - 244,28.
Молекулярная масса BaSO4 - 233,4.
Расчет титра сводится к следующему:
244,28
---------------
233,4
![]() (г)
(г)
---------------
10 (г)
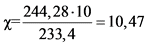 г
г
Таким образом, для приготовления 100 мл раствора BaCl2 с титром BaSO4 ~ 100 мг/мл необходима навеска соли (BaCl2·2H2O) массой 10,47 г. Навеску помещают в стакан емкостью 150 мл, растворяют в дистиллированной воде, добавляют 5 - 10 мл 2 н HCl. Раствор переливают в мерную колбу емкостью 100 мл (если необходимо - фильтруют), доводят дистиллированной водой до метки.
Определение титра раствора проводят в условиях, аналогичных тем, которые используются на последнем этапе радиохимического анализа при выделении изотопов радия на BaSO4.
Для этого в пять стаканов емкостью 150 мл отбирают по 1 мл приготовленного раствора, добавляют 10 - 15 мл 2 н HCl и разбавляют дистиллированной водой до 100 мл. Нагревают раствор до кипения и осаждают BaSO4, приливая при непрерывном перемешивании 10 мл 10% H2SO4. После отстаивания в течение 30 - 40 минут осадки фильтруют через обеззоленный фильтр "синяя лента", тщательно перенося их со стенок стаканов горячей дистиллированной водой. Фильтры с осадками помещают в тигли, обугливают на электрической плитке, переносят в муфельную печь, прокаливают при 800 °C в течение 1 часа, после охлаждения взвешивают. Вычисляют среднее арифметическое значение массы осадка, которое считают точной концентрацией, т.е. титром бария (![]() ) в растворе.
) в растворе.
2. Приготовление титрованного раствора лантана
Исходная соль для приготовления раствора - LaCl3·7H2O. Соединение, в виде которого выделяется носитель на последней стадии анализа, - La2O3. Объем приготовляемого раствора носителя - 100 мл. Заданный титр La2O3 - 50 мг/мл, т.е. в 100 мл раствора должно содержаться 50 мг x 100 мл = 5 г La2O3.
Молекулярная масса LaCl3·7H2O - 371,38.
Молекулярная масса La2O3 - 325,82.
Для получения одной молекулы La2O3 необходимо две молекулы LaCl3·7H2O, молекулярная масса которых составляет 742,46.
Расчет титра сводится к следующему:
742,46
---------------
325,82
![]() (г)
(г)
---------------
5 (г)
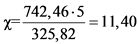 г
г
Таким образом, для приготовления 100 мл раствора LaCl3 с титром La2O3 ~ 50 мг/мл необходима навеска соли (LaCl3·7H2O) массой 11,40 г. Навеску помещают в стакан емкостью 150 мл, растворяют при нагревании в 20 - 40 мл 2 н HCl. После охлаждения раствор переливают в мерную колбу емкостью 100 мл, доводят дистиллированной водой до метки.
Для определения титра приготовленного раствора используют приемы, указанные в методике выделения 228Ac на La(OH)3 на последнем этапе его радиохимического анализа. Для этого в пять стаканов емкостью 150 мл отбирают по 1 мл приготовленного раствора носителя, добавляют 10 мл 2 н HCl и разбавляют дистиллированной водой до 100 мл. Нагревают раствор до 80 °C и осаждают La(OH)3, добавляя раствор NH4OH до pH = 8 - 9. Осадки фильтруют через обеззоленный фильтр "белая лента", тщательно перенося их со стенок стаканов горячей дистиллированной водой. Фильтры с осадками помещают в тигли, обугливают на электрической плитке, переносят в муфельную печь, прокаливают при 800 °C в течение 1 часа, после охлаждения взвешивают. Вычисляют среднее арифметическое значение массы осадка, которое считают точной концентрацией, т.е. титром лантана (![]() ) в растворе.
) в растворе.
3. Приготовление титрованного раствора железа
Исходная соль для приготовления раствора - FeCl3·6H2O. Соединение, в виде которого выделяется носитель на последней стадии анализа, - Fe2O3. Объем приготовляемого раствора носителя - 100 мл. Заданный титр Fe2O3 - 20 мг/мл, т.е. в 100 мл раствора должно содержаться 20 мг x 100 мл = 2 г La2O3.
Молекулярная масса FeCl3·6H2O - 270,3.
Молекулярная масса Fe2O3 - 159,69.
Для получения одной молекулы Fe2O3 необходимо две молекулы FeCl3·6H2O, молекулярная масса которых составляет 540,6.
Расчет титра сводится к следующему:
540,6
---------------
159,69
![]() (г)
(г)
---------------
2 (г)
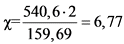 г
г
Таким образом, для приготовления 100 мл раствора FeCl3 с титром Fe2O3 ~ 20 мг/мл необходима навеска соли (FeCl3·6H2O) массой 6,77 г. Навеску помещают в стакан емкостью 150 мл, добавляют 10 мл 2 н HCl и дистиллированную воду до 80 мл. Раствор переливают в мерную колбу емкостью 100 мл и доводят дистиллированной водой до метки.
Для определения титра раствора в пять стаканов емкостью 150 мл отбирают по 1 мл приготовленного раствора носителя, добавляют 10 мл 2 н HCl и разбавляют дистиллированной водой до 100 мл. Нагревают раствор до 80 °C и осаждают Fe(OH)3, добавляя раствор NH4OH до pH = 8 - 9. Осадки фильтруют через обеззоленный фильтр "белая лента", тщательно перенося их со стенок стаканов горячей дистиллированной водой. Фильтры с осадками помещают в тигли, обугливают на электрической плитке, переносят в муфельную печь, прокаливают при 800 °C в течение 1 часа, после охлаждения взвешивают. Вычисляют среднее арифметическое значение массы осадка, которое считают точной концентрацией, т.е. титром железа (![]() ) в растворе.
) в растворе.
4. Приготовление титрованного раствора иттрия
Исходная соль для приготовления раствора - YCl3·6H2O. Соединение, в виде которого выделяется носитель на последней стадии анализа, - Y2O3. Объем приготовляемого раствора носителя - 100 мл. Заданный титр Y2O3 - 60 мг/мл, т.е. в 100 мл раствора должно содержаться 60 мг x 100 мл = 6 г Y2O3.
Молекулярная масса YCl3·6H2O - 303,4.
Молекулярная масса Y2O3 - 225,8.
Для получения одной молекулы Y2O3 необходимо две молекулы YCl3·6H2O, молекулярная масса которых составляет 606,8.
Расчет титра сводится к следующему:
606,8
---------------
225,8
![]() (г)
(г)
---------------
6 (г)
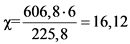 г
г
Таким образом, для приготовления 100 мл раствора YCl3 с титром Y2O3 ~ 60 мг/мл необходима навеска соли (YCl3·6H2O) массой 16,12 г. Навеску помещают в стакан емкостью 150 мл, добавляют 15 - 20 мл 2 н HCl, нагревают до растворения осадка, разбавляют дистиллированной водой до 70 мл, охлаждают. Раствор переливают в мерную колбу емкостью 100 мл и доводят дистиллированной водой до метки.
Для определения титра раствора в пять стаканов емкостью 150 мл отбирают по 1 мл приготовленного раствора носителя, добавляют 10 - 15 мл 2 н HCl и дистиллированную воду до 60 - 70 мл. После нагревания до 70 °C добавляют 10 мл насыщенного раствора H2C2O4 и раствором и NH4OH доводят до pH ~ 1,5. После отстаивания (около 30 минут) осадки фильтруют через обеззоленный фильтр "синяя лента", промывают дистиллированной водой с добавлением H2C2O4. Фильтры с осадками помещают в тигли, обугливают на электрической плитке, переносят в муфельную печь, прокаливают при 800 °C в течение 1 часа, после охлаждения взвешивают. Вычисляют среднее арифметическое значение массы осадка, которое считают точной концентрацией, т.е. титром иттрия (![]() ) в растворе.
) в растворе.
5. Приготовление титрованного раствора стронция
Исходная соль для приготовления раствора - SrCl2·6H2O. Соединение, в виде которого выделяется носитель на последней стадии анализа - SrSO4. Объем приготовляемого раствора носителя - 100 мл. Заданный титр SrSO4 - 100 мг/мл, т.е. в 100 мл раствора должно содержаться 100 мг x 100 мл = 10 г SrSO4.
Молекулярная масса SrCl2·6H2O - 266,62.
Молекулярная масса SrSO4 - 183,7.
Расчет титра сводится к следующему:
266,62
---------------
183,7
![]() (г)
(г)
---------------
10 (г)
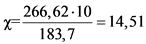 г
г
Таким образом, для приготовления 100 мл раствора SrCl2 с титром SrSO4 ~ 100 мг/мл необходима навеска соли (SrCl2·6H2O) массой 14,51 г. Навеску помещают в стакан емкостью 150 мл, добавляют 30 мл 2 н HCl, разбавляют дистиллированной водой до 70 мл. Нагревают до полного растворения осадка. После охлаждения раствор переносят в мерную колбу емкостью 100 мл, доводят дистиллированной водой до метки.
Для определения титра в пять стаканов емкостью 150 мл отбирают по 1 мл приготовленного раствора, добавляют 10 - 15 мл 2 н HCl, 10 - 15 мл дистиллированной воды, 10 мл 10% H2SO4 и равное по объему количество этилового спирта. После отстаивания в течение 2 - 3 часов осадки фильтруют через обеззоленный фильтр "синяя лента", тщательно перенося их со стенок стаканов горячей дистиллированной водой с добавлением нескольких капель 10% H2SO4. Фильтры с осадками помещают в тигли, обугливают на электрической плитке, переносят в муфельную печь, прокаливают при 800 °C в течение 1 часа, после охлаждения взвешивают. Вычисляют среднее арифметическое значение массы осадка, которое считают точной концентрацией, т.е. титром стронция (![]() ) в растворе.
) в растворе.
6. Приготовление титрованного раствора цезия
Исходная соль для приготовления раствора - CsCl. Соединение, в виде которого выделяется носитель на последней стадии анализа - Cs3Sb2I9. Объем приготовляемого раствора носителя - 100 мл. Заданный титр Cs3Sb2I9 - 100 мг/мл, т.е. в 100 мл раствора должно содержаться 100 мг x 100 мл = 10 г Cs3Sb2I9.
Молекулярная масса CsCl - 168,36.
Молекулярная масса Cs3Sb2I9 - 1784,2.
Для получения одной молекулы Cs3Sb2I9 необходимо три молекулы CsCl, молекулярная масса которых составляет 505,08.
Расчет титра сводится к следующему:
505,08
---------------
1784,2
![]() (г)
(г)
---------------
10 (г)
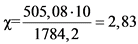 г
г
Таким образом, для приготовления 100 мл раствора CsCl с титром Cs3Sb2I9 ~ 100 мг/мл необходима навеска соли CsCl массой 2,83 г. Навеску помещают в стакан емкостью 150 мл, добавляют 70 - 80 мл дистиллированной воды, 5 мл 2 н HCl, переливают в мерную колбу емкостью 100 мл и доводят дистиллированной водой до метки.
Для определения титра раствора в пять стаканов емкостью 150 мл отбирают по 1 мл приготовленного раствора носителя, 30 - 40 мл 3 н HCl, 3 мл свежеприготовленного раствора NH4I или 3,5 мл KI (170 г NH4I на 100 мл H2O; 140 г KI на 100 мл H2O), затем при постоянном перемешивании добавляют 0,5 - 0,8 мл раствора SbCl3 до выпадения осадка Cs3Sb2I9 красного цвета. Осадок выдерживают 1 - 2 часа на водяной бане, фильтруют через обеззоленный фильтр "синяя лента", промывают 3 раза ледяной уксусной кислотой, этиловым спиртом, сушат под инфракрасной лампой, взвешивают. Вычисляют среднее арифметическое значение массы осадка, которое считают точной концентрацией, т.е. титром цезия (![]() ) в растворе.
) в растворе.
Подготовка ионообменных смол к анализу, регенерация и хранение
Для удовлетворительного хроматографического разделения ионов следует пользоваться только мелкозернистыми ионитами. Размер зерен смол (ЭДЭ-10П, КУ-2-8), используемых для разделения урана и тория - (0,125 ![]() 0,250) мм.
0,250) мм.
Перед использованием ионит просеивают через стандартные сита для того, чтобы сразу отделить нужную фракцию. Крупные зерна ионита измельчают в ступке мокрым или воздушно-сухим способом. После этого ионит снова просеивают, а оставшиеся крупные зерна измельчают.
Подготовленной таким образом фракции ионита нужно дать набухнуть, причем в процессе набухания следует сливать верхнюю жидкость для удаления из смолы частиц почти коллоидного размера. Для этого в мерном цилиндре приготавливают взвесь смолы в десятикратном объеме воды и оставляют ее приблизительно на полчаса. Образовавшуюся над смолой облакообразную взвесь сливают, добавляют в цилиндр порцию воды и повторяют весь процесс до тех пор, пока слой жидкости над смолой не станет чистым. В течение всего этого процесса смола впитывает воду и набухает. Далее проводят подготовительный цикл, при котором ионит переводят в Cl--форму: смолы обрабатывают 1 - 2 н раствором HCl, дистиллированной водой, 0,5 - 1 н раствором NaOH и снова дистиллированной водой.
Для переведения смолы в Cl--форму ее обрабатывают 1 н раствором HCl и промывают дистиллированной водой. Каждый раз смолу нужно промывать дистиллированной водой до нейтральной реакции. Время обработки смолы каждым реактивом - несколько часов, при переведении в Cl--форму смолу, залитую 1 н раствором HCl, лучше оставить на ночь.
По окончании анализа смолу переносят из колонок в стакан, промывают дистиллированной водой, сливают верхнюю жидкость и проводят полный подготовительный цикл.
После регенерации смолу тщательно промывают дистиллированной водой и хранят в закрытых сосудах под слоем дистиллированной воды. Смолу, которая хранилась в течение года или дольше, перед использованием рекомендуется вновь регенерировать [5, 6].
Приложение 5
к МР 2.6.1.0064-12
(справочное)
ПРИГОТОВЛЕНИЕ
КАЛИБРОВОЧНЫХ ОБРАЗЦОВ И ГРАДУИРОВКА ![]() -
-![]() РАДИОМЕТРА
РАДИОМЕТРА
1. Подготовка аппаратуры к измерениям
1.1. Измерения активности радионуклидов выполняют с применением ![]() -
-![]() радиометра УМФ-2000.
радиометра УМФ-2000.
1.2. Проводят подготовку радиометра к работе согласно Руководству по эксплуатации на прибор.
1.3. Устанавливают в барабан радиометра чистую, пустую измерительную кювету и выполняют измерение фона в ![]() -канале и
-канале и ![]() -канале в течение не менее 60 минут.
-канале в течение не менее 60 минут.
1.4. Измерения выполняют в соответствии с Техническим описанием и инструкцией по эксплуатации радиометра УМФ-2000.
2. Градуировка ![]() -
-![]() радиометра для измерения активности
радиометра для измерения активности ![]() -излучающих радионуклидов (226Ra, 224Ra) и суммарной
-излучающих радионуклидов (226Ra, 224Ra) и суммарной ![]() -активности
-активности
Коэффициенты связи между активностью и скоростью счета импульсов от счетного образца ![]() -излучающих радионуклидов для толстослойных препаратов, имеющих поверхностную плотность более 10 мг/см2, (Бк/мг/(имп./мин.), определяют с использованием эталонного раствора 226Ra с удельной объемной активностью 10 - 100 Бк/см3.
-излучающих радионуклидов для толстослойных препаратов, имеющих поверхностную плотность более 10 мг/см2, (Бк/мг/(имп./мин.), определяют с использованием эталонного раствора 226Ra с удельной объемной активностью 10 - 100 Бк/см3.
Для приготовления калибровочных образцов используют аликвоты эталонного раствора (готовят 5 образцов) 226Ra одинаковой активности. В 5 термостойких химических стаканов емкостью 100 мл приливают по 30 - 40 мл 2 н HCl и вносят раствор носителя 226Ra - хлористый барий BaCl2 (90 - 100 мг в расчете на BaSO4). В полученные растворы вносят отобранные аликвоты эталонного раствора 226Ra одинаковой активности. Приготовленные растворы кипятят 10 - 15 минут на электрической плитке, добавляют при непрерывном перемешивании 15 мл 10% H2SO4, кипятят 10 - 15 мин., после чего осадку (Ba(Ra)SO4) дают отстояться в течение 1 часа. Осадок отфильтровывают через обеззоленный фильтр "синяя лента" (масса золы одного фильтра - 0,0004 г) и несколько раз промывают дистиллированной водой. Промытый осадок сульфата бария Ba(Ra)SO4 высушивают, прокаливают в муфельной печи при температуре 800 °C в течение 1 часа, взвешивают, растирают в тигле стеклянной палочкой до состояния "пудры" и переносят шпателем на предварительно взвешенную подложку для измерения активности 226Ra. Смачивают перенесенный на подложку осадок несколькими каплями этилового спирта, с помощью стеклянной палочки равномерно распределяют и уплотняют на подложке, затем высушивают под инфракрасной лампой. Высушенный осадок взвешивают (вместе с подложкой) для определения массы полученного образца. Через 4 часа после прокаливания проводят измерение полученных образцов на УМФ-2000. Вычисляют скорость счета импульсов от счетных образцов по ![]() -каналу. Допустимо расхождение в значениях скорости счета импульсов от приготовленных образцов не более 5%. Скорость счета импульсов от 226Ra берется как среднее значение данной величины от 5 приготовленных образцов.
-каналу. Допустимо расхождение в значениях скорости счета импульсов от приготовленных образцов не более 5%. Скорость счета импульсов от 226Ra берется как среднее значение данной величины от 5 приготовленных образцов.
Рассчитывают коэффициент связи (KRa-226) между активностью и скоростью счета импульсов от излучения 226Ra в толстослойном образце по формуле:
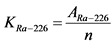 , (Бк/мг)/(имп./мин.), (31)
, (Бк/мг)/(имп./мин.), (31)
где:
ARa-226 - массовая активность толстослойного градуировочного образца (BaSO4), приготовленного из отобранной аликвоты эталонного раствора, Бк/мг;
n - средняя скорость счета от серии приготовленных толстослойных образцов BaSO4, имп./мин.
Полученный коэффициент связи между активностью и скоростью счета импульсов от излучения 226Ra в счетных образцах используется только для расчетов активности ![]() -излучения толстослойных образцов, измеренных в идентичных условиях:
-излучения толстослойных образцов, измеренных в идентичных условиях:
- калибровочные и счетные образцы, приготовленные для измерения, должны иметь одинаковые размеры;
- геометрия измерения указанных образцов должна быть одинаковой.
При изменении условий измерения, например при использовании подложек иной площади, процедуру калибровки повторяют, применяя для приготовления счетных образцов соответствующие подложки.
Приготовленный эталон можно использовать для проверки коэффициента связи в течение длительного времени. По прошествии 36 суток от его первичного измерения значение коэффициента связи рассчитывают по формуле:
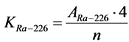 , (Бк/мг)/(имп./мин.) (32)
, (Бк/мг)/(имп./мин.) (32)
Расчет коэффициента связи для 224Ra проводят по коэффициенту для 226Ra, определенному согласно Приложению 5, учитывая несоответствие их энергетических спектров по формуле:
KRa-224 = 0,77·KRa-226, (Бк/мг)/(имп./мин.) (33)
3. Градуировка ![]() -
-![]() радиометра для измерения активности 210Po и 210Bi(210Pb)
радиометра для измерения активности 210Po и 210Bi(210Pb)
Градуировка ![]() -
-![]() радиометра для измерения тонкослойных препаратов 210Po и 210Bi(210Pb) производится с помощью контрольного образца сравнения, в качестве которого используют диск из нержавеющей стали диаметром 34 мм и площадью 5 см2, на который методом электролитического осаждения нанесены эталонные растворы указанных радионуклидов.
радиометра для измерения тонкослойных препаратов 210Po и 210Bi(210Pb) производится с помощью контрольного образца сравнения, в качестве которого используют диск из нержавеющей стали диаметром 34 мм и площадью 5 см2, на который методом электролитического осаждения нанесены эталонные растворы указанных радионуклидов.
Измерение активности радионуклидов 210Po, 210Bi, 210Pb в контрольном образце сравнения проводится в соответствии с пунктом 13.5.
Коэффициент связи (KPo-210) между ![]() -активностью 210Po в контрольном образце сравнения и скоростью счета импульсов от
-активностью 210Po в контрольном образце сравнения и скоростью счета импульсов от ![]() -излучения 210Po в этом образце (Бк/(имп./мин.) определяют по формуле:
-излучения 210Po в этом образце (Бк/(имп./мин.) определяют по формуле:
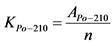 , Бк/(имп./мин.), (34)
, Бк/(имп./мин.), (34)
где:
APo-210 - активность 210Po в контрольном образце сравнения, Бк;
n - скорость счета импульсов за вычетом скорости счета фона, имп./мин.
Коэффициент связи (KBi-210) между ![]() -активностью 210Bi в контрольном образце сравнения и скоростью счета импульсов от
-активностью 210Bi в контрольном образце сравнения и скоростью счета импульсов от ![]() -излучения 210Bi в этом образце определяют по формуле:
-излучения 210Bi в этом образце определяют по формуле:
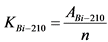 , Бк/(имп./мин.), (35)
, Бк/(имп./мин.), (35)
где:
ABi-210 - активность 210Bi в контрольном образце сравнения, Бк;
n - скорость счета импульсов за вычетом скорости счета фона, имп./мин.
4. Градуировка ![]() -
-![]() радиометра для измерения активности
радиометра для измерения активности ![]() -излучающих радионуклидов: 40K, 137Cs, 90Sr(90Y), 228Ra(228Ac), 234Th(234Pa) и суммарной
-излучающих радионуклидов: 40K, 137Cs, 90Sr(90Y), 228Ra(228Ac), 234Th(234Pa) и суммарной ![]() -активности
-активности
4.1. Для приготовления калибровочных образцов 40K используется химически чистый хлористый калий, удельная ![]() -активность которого равна 0,0143 Бк/мг. KCl предварительно прокаливают при температуре 120 - 130 °C в течение 2 часов, затем растирают в фарфоровой ступке. Готовят серию из 10 - 15 контрольных источников из разных навесок KCl (от 20 до 200 мг) на таких же подложках, какие используются для радиометрии счетных образцов определяемых радионуклидов. Активность 40K в приготовленных счетных образцах измеряют в соответствии с п. 13.10.
-активность которого равна 0,0143 Бк/мг. KCl предварительно прокаливают при температуре 120 - 130 °C в течение 2 часов, затем растирают в фарфоровой ступке. Готовят серию из 10 - 15 контрольных источников из разных навесок KCl (от 20 до 200 мг) на таких же подложках, какие используются для радиометрии счетных образцов определяемых радионуклидов. Активность 40K в приготовленных счетных образцах измеряют в соответствии с п. 13.10.
Рассчитывают коэффициенты связи между активностью счетных образцов разной массы и скоростью счета импульсов от излучения 40K по формуле:
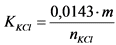 , Бк/(имп./мин.), (36)
, Бк/(имп./мин.), (36)
где:
0,0143 - удельная ![]() -активность KCl, Бк/мг;
-активность KCl, Бк/мг;
m - масса измеряемого препарата, мг;
nKCl - скорость счета импульсов (имп./мин.) от препарата KCl массой m, мг.
Строят график зависимости коэффициентов связи от массы счетного образца на подложке (Рис. 1).
Примечание:
Зная коэффициент связи между активностью и скоростью счета импульсов KCl, можно определить коэффициент связи для бета-излучающих радионуклидов со средней энергией излучения не менее 1000 кэВ (![]() ):
):
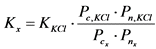 , Бк/(имп./мин.) (37)
, Бк/(имп./мин.) (37)
Коэффициент самопоглощения (Pс) определяют по формуле:
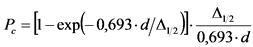 , (38)
, (38)
где:
d - толщина слоя препарата на подложке, мг/см2;
![]() - слой половинного ослабления
- слой половинного ослабления ![]() -частиц, мг/см2.
-частиц, мг/см2.
Поправку на поглощение ![]() -частиц в материале окошка счетчика определяют по формуле:
-частиц в материале окошка счетчика определяют по формуле:
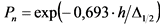 , (39)
, (39)
где: h - толщина материала окошка счетчика, мг/см2.
4.2. Для приготовления калибровочных образцов 90Sr(90Y) используется эталонный раствор радионуклидов 90Sr(90Y) удельной объемной активностью 10 - 100 Бк/см3. К отобранным аликвотам эталонного раствора одинакового объема, перенесенным в термостойкие химические стаканы (готовят 10 - 15 образцов), добавляют 20 - 30 мл 2 н HCl, вносят растворы носителей 90Y - хлористый иттрий (от 20 до 200 мг в расчете на Y2O3) и хлористый стронций (100 мг в расчете на SrSO4), разбавляют дистиллированной водой до 100 мл и кипятят в течение 20 - 30 минут. Из горячего раствора осаждают гидроокись иттрия добавлением безугольного аммиака до pH = 8 - 9, осадок отфильтровывают и промывают горячей дистиллированной водой. Время отделения 90Y от 90Sr записывают и учитывают при расчете активности. Осадок переносят в фарфоровый тигель и прокаливают в муфеле при температуре 800 - 900 °C до получения окиси иттрия Y2O3. Полученный осадок наносят равномерным слоем на предварительно взвешенную подложку, взвешивают осадок вместе с подложкой для определения массы полученного образца. Таким образом, готовят 10 - 15 образцов с разной массой для измерения активности 90Sr по его дочернему радионуклиду 90Y. Активность 90Y в приготовленном счетном образце измеряют в соответствии с п. 13.8.
Рассчитывают коэффициенты связи между активностью счетных образцов разной массы и скоростью счета импульсов от излучения 90Y по формуле:
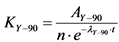 , (Бк)/(имп./мин.), (40)
, (Бк)/(имп./мин.), (40)
где:
AY-90 - активность 90Y в счетном образце, Бк;
n - скорость счета импульсов в счетном образце за вычетом скорости счета фона, имп./мин.;
![]() - поправка на распад 90Y, (Таблица 6).
- поправка на распад 90Y, (Таблица 6).
Строят график зависимости коэффициентов связи от массы счетного образца на подложке (Рис. 1).
4.3. Для приготовления калибровочных образцов 137Cs используют эталонный раствор радионуклида 137Cs удельной объемной активностью 10 - 100 Бк/см3. В стаканы на 50 мл вносят растворы носителя 137Cs - хлористый цезий (от 20 до 200 мг в расчете на Cs3Sb2I9) и аликвоты эталонного раствора одинакового объема, добавляют 30 - 40 мл 3 н HCl и кипятят 15 - 20 минут. После чего раствор разбавляют дистиллированной водой до первоначального объема и охлаждают. В полученные растворы вносят по 3 мл свежеприготовленного насыщенного раствора NH4I (или KI), приливают 0,8 мл 10% раствора треххлористой сурьмы SbCl3 и хорошо перемешивают раствор стеклянной палочкой до образования красного осадка. Осадок выдерживают в течение 1,5 - 2 часа (можно на ледяной бане). Отстоявшийся осадок Cs3Sb2I9 фильтруют, промывают 3 - 4 раза ледяной уксусной кислотой (до осветления промывных вод), затем этиловым спиртом до удаления запаха уксусной кислоты и высушивают. Высушенный осадок наносят равномерным слоем на предварительно взвешенную подложку, взвешивают осадок вместе с подложкой для определения массы полученного образца. Таким образом готовят 10 - 15 образцов с разной массой для измерения активности 137Cs. Активность 137Cs в приготовленных счетных образцах измеряют в соответствии с п. 13.9.
Рассчитывают коэффициенты связи между активностью счетных образцов разной массы и скоростью счета импульсов от излучения 137Cs по формуле:
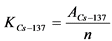 , (Бк)/(имп./мин.), (41)
, (Бк)/(имп./мин.), (41)
где:
ACs-137 - активность 137Cs в счетном образце, Бк;
n - скорость счета импульсов в счетном образце за вычетом скорости счета фона, имп./мин.
Строят график зависимости коэффициентов связи от массы счетного образца на подложке (Рис. 1).
Рис. 1. Зависимость коэффициентов связи между
скоростью счета импульсов от ![]() -излучения толстослойных
-излучения толстослойных
препаратов радионуклидов и их активностью от массы счетного
образца на подложке площадью 2,5 см2 (не приводится)
4.4. Для бета-излучающих радионуклидов - 228Ac, находящегося в радиоактивном равновесии с 228Ra, и 234Pa, находящегося в радиоактивном равновесии с 234Th, можно использовать значения коэффициентов связи между скоростью счета импульсов и активностью радионуклидов, полученные для 90Y, т.к. максимальные энергии ![]() -излучения указанных радионуклидов различаются незначительно (234Pa - 2300 кэВ, 228Ac - 2089 кэВ, 90Y - 2274 кэВ).
-излучения указанных радионуклидов различаются незначительно (234Pa - 2300 кэВ, 228Ac - 2089 кэВ, 90Y - 2274 кэВ).
Приложение 6
к МР 2.6.1.0064-12
(справочное)
СХЕМА
Радиохимическое определение удельной активности природных
радионуклидов в воде (224Ra, 226Ra, 228Ra, 234U, 238U,
210Po, 210Pb, 210Bi, 228Th, 230Th, 232Th)
Схема
радиохимического определения удельной активности
90Sr и 137Cs в воде
Приложение 7
к МР 2.6.1.0064-12
ПЕРЕЧЕНЬ МЕТОДИК РАДИАЦИОННОГО КОНТРОЛЯ ПИТЬЕВОЙ ВОДЫ
1. Отбор и подготовка проб питьевой воды для определения показателей радиационной безопасности. Методические рекомендации, утв. Роспотребнадзором от 27.12.2007 N 0100/13609-07-34.
2. Радиационный контроль и гигиеническая оценка источников питьевого водоснабжения и питьевой воды по показателям радиационной безопасности. Методические указания МУ 2.6.1.1981-05. - М.: 2005.
3. ГОСТ Р 51730-2001. Вода питьевая. Метод определения суммарной удельной активности радионуклидов.
4. Суммарная активность альфа- и бета-излучающих радионуклидов в природных водах (пресных и минерализованных). Подготовка проб и измерения. Методические рекомендации. Москва, ВИМС, 2009. Утв. ЦМИИ ФГУП ВНИИФТРИ Ростехрегулирование, 2009.
5. Методика выполнения измерений объемной активности полония-210 и свинца-210 в природных водах альфа-, бета-радиометрическим методом с радиохимической подготовкой. Свидетельство ЦМИИ ГНМЦ ВНИИФТРИ Госстандарта РФ N 49090.3Н618 от 18.12.2003; Свидетельство НСАМ N 396-ЯФ, Москва, ВИМС, 2001 - 2003.
6. Методика выполнения измерений объемной активности изотопов урана (234, 238) в природных водах с минерализацией до 5 г/дм3 альфа-спектрометрическим методом с радиохимической подготовкой. Свидетельство ЦМИИ ГНМЦ ВНИИФТРИ Госстандарта РФ N 49090.3Н628 от 18.12.2003. Свидетельство НСАМ N 381-ЯФ, Москва, ВИМС, 2003.
7. Методика выполнения измерений объемной активности изотопов тория (232, 230, 228) в природных водах с минерализацией до 5 г/дм3 альфа-спектрометрическим методом с радиохимической подготовкой. Свидетельство ЦМИИ ГНМЦ ВНИИФТРИ Госстандарта РФ N 49090.3Н625 от 18.12.2003; Свидетельство НСАМ N 461-ЯФ, Москва, ФГУП ВИМС, 2003.
8. Методика выполнения измерений объемной активности изотопов радия (226, 228) в пробах природных вод с минерализацией до 5 г/дм3 альфа-, бета-радиометрическим методом с радиохимической подготовкой. Свидетельство ЦМИИ ГНМЦ ВНИИФТРИ Госстандарта РФ N 40090.6Б327 от 28.02.2006, Москва, ВИМС, 2000.
9. Методика радиохимического приготовления счетных образцов из проб питьевой воды для измерения активности Po-210, общей альфа-активности (без Po-210) и общей бета-активности (без K-40) на радиологическом комплексе с программным обеспечением "Прогресс". Свидетельство об аттестации методики ФГУП ВНИИФТРИ N 42090.6В525 от 27.03.2006.
10. Методика приготовления счетных образцов из проб питьевой воды для измерения активности ЕРН с использованием радиологического комплекса с программным обеспечением "Прогресс". Свидетельство об аттестации методики ФГУП ВНИИФТРИ N 42090.6В524 от 27.03.2006.
11. Методика выполнения измерений объемной активности изотопов плутония (239 + 240, 238) в природных водах с минерализацией до 5 г/дм3 альфа-спектрометрическим методом с радиохимической подготовкой. Свидетельство ЦМИИ ГНМЦ ВНИИФТРИ Госстандарта РФ N 49090.3Н622 от 18.12.2003; Свидетельство НСАМ N 407-ЯФ, Москва, ВИМС, 1999.
12. Методические рекомендации по определению естественных изотопов: радия-224, свинца-210, тория-232, урана-238, радия-226 в пробах питьевой воды, почвы и золы растений. МР ЛНИИРГ МЗ РСФСР. Л., 1978.
13. Методические рекомендации по санитарному контролю за содержанием радиоактивных веществ в объектах внешней среды. Под ред. А.Н. Марея и А.С. Зыковой. М., МЗ СССР, 1980, 88 с.
14. Методика выполнения измерений удельной активности радионуклидов 238U, 234U, 232Th, 230Th, 228Th, 228Ra, 226Ra, 224Ra, 210Pb, 210Po, 40K, 137Cs, 90Sr и суммарной удельной активности альфа-, бета-излучающих радионуклидов в воде с применением альфа-, бета-радиометра и альфа-спектрометра. Разработана ФГУН НИИРГ имени профессора П.В. Рамзаева Роспотребнадзора. Свидетельство ФГУП "ВНИИМ им. Д.И. Менделеева" Федерального государственного агентства по техническому регулированию и метрологии N 1212/07 от 26 октября 2007 г.
15. Методика выполнения измерений удельной активности радионуклидов уран-234, уран-238 в воде с применением полупроводникового альфа-спектрометра в воде. Разработана ФГУН НИИРГ имени профессора П.В. Рамзаева Роспотребнадзора. Свидетельство ФГУП "ВНИИМ им. Д.И. Менделеева" Федерального государственного агентства по техническому регулированию и метрологии N 207/08 от 29 февраля 2008 г.
Приложение 8
к МР 2.6.1.0064-12
(рекомендуемое)
ИНФОРМАЦИЯ,
КОТОРАЯ ДОЛЖНА БЫТЬ ОТРАЖЕНА В ПРОТОКОЛЕ ИСПЫТАНИЙ
N п/п
Наименование средств измерений
Заводской номер
N свидетельства о госповерке, дата
Дата окончания действия свидетельства
Кем выдано свидетельство
N п/п
Определяемый показатель
Результат измерения
Неопределенность измерения
Гигиенический критерий (КУ, УВ)
Единицы измерения
![]()
0,2
Бк/кг
![]()
1,0
Бк/кг
210Po
0,11
Бк/кг
...
Бк/кг
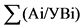 <*>
<*>
1,0
-
СПИСОК ИСПОЛЬЗОВАННОЙ ЛИТЕРАТУРЫ
1. Ю.А. Сапожников, Р.А. Алиев, С.Н. Калмыков. Радиоактивность окружающей среды. М.: БИНОМ. Лаборатория знаний, 2006. - 286 с.: ил. - (Методы в химии);
2. Аналитическая химия урана. Главный редактор А.Н. Виноградов. М.: Академия наук СССР, 1962. - 430 с.;
3. В.М. Вдовенко, Ю.В. Дубасов. Аналитическая химия радия. Л.: Наука, 1973. - 189 с.;
4. Методические рекомендации по санитарному контролю за содержанием радиоактивных веществ в объектах внешней среды. Под ред. А.Н. Марея и А.С. Зыковой. М., МЗ СССР, 1980, 88 с.;
5. Краткий справочник химика. Ред. Б.В. Некрасов. М.: Государственное научно-техническое издательство химической литературы, 1954. - 559 с.;
6. О.О. Самуэльсон. Ионообменные разделения в аналитической химии. М.-Л.: Химия, 1966;
7. Жидкостная колоночная хромотография. Под редакцией З. Дейла. М.: Мир, т. 1, 1978.