Решение Совета Евразийской экономической комиссии от 12.04.2024 N 30
СОВЕТ ЕВРАЗИЙСКОЙ ЭКОНОМИЧЕСКОЙ КОМИССИИ
РЕШЕНИЕ
от 12 апреля 2024 г. N 30
О ВНЕСЕНИИ ИЗМЕНЕНИЙ
В ПРАВИЛА ПРОВЕДЕНИЯ ИССЛЕДОВАНИЙ БИОЭКВИВАЛЕНТНОСТИ
ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ В РАМКАХ ЕВРАЗИЙСКОГО
ЭКОНОМИЧЕСКОГО СОЮЗА
В соответствии с пунктом 2 статьи 4 и статьей 6 Соглашения о единых принципах и правилах обращения лекарственных средств в рамках Евразийского экономического союза от 23 декабря 2014 года и пунктом 86 приложения N 1 к Регламенту работы Евразийской экономической комиссии, утвержденному Решением Высшего Евразийского экономического совета от 23 декабря 2014 г. N 98, Совет Евразийской экономической комиссии решил:
1. Внести в Правила проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза, утвержденные Решением Совета Евразийской экономической комиссии от 3 ноября 2016 г. N 85, изменения согласно приложению.
2. Настоящее Решение вступает в силу по истечении 30 календарных дней с даты его официального опубликования.
Члены Совета Евразийской экономической комиссии:
От Республики Армения
М.ГРИГОРЯН
От Республики Беларусь
И.ПЕТРИШЕНКО
От Республики Казахстан
С.ЖУМАНГАРИН
От Кыргызской Республики
А.КАСЫМАЛИЕВ
От Российской Федерации
А.ОВЕРЧУК
Приложение
к Решению Совета
Евразийской экономической комиссии
от 12 апреля 2024 г. N 30
ИЗМЕНЕНИЯ
ВНОСИМЫЕ В ПРАВИЛА ПРОВЕДЕНИЯ ИССЛЕДОВАНИЙ
БИОЭКВИВАЛЕНТНОСТИ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ В РАМКАХ
ЕВРАЗИЙСКОГО ЭКОНОМИЧЕСКОГО СОЮЗА
1. По тексту пункта 10 слова "при Комиссии" исключить.
2. Пункт 18 изложить в следующей редакции:
"18. При выборе референтного лекарственного препарата исходят из следующей последовательности:
а) оригинальный лекарственный препарат, безопасность, эффективность и качество которого были установлены при регистрации в Союзе, а также зарегистрированный в соответствии с законодательством государств-членов ("зарегистрированный в Союзе оригинальный препарат");
б) оригинальный лекарственный препарат, зарегистрированный в странах региона Международного совета по гармонизации технических требований к лекарственным препаратам для медицинского применения (ICH), либо при обосновании его отсутствия в обращении на фармацевтическом рынке стран региона ICH или невозможности его приобретения воспроизведенный (или гибридный) лекарственный препарат, зарегистрированный как минимум в одном из государств-членов и подтвердивший свою биоэквивалентность оригинальному лекарственному препарату (при одобрении выбора воспроизведенного препарата Экспертным комитетом), при невозможности выполнения подпункта "а" настоящего пункта;
в) лекарственные препараты, утвержденные Экспертным комитетом (список решений размещен на официальном сайте Союза), при невозможности выполнения подпунктов "а" и "б" настоящего пункта;
г) лекарственный препарат, имеющий опыт применения на территории как минимум одного из государств-членов не менее 20 лет (при одобрении Экспертным комитетом), при невозможности выполнения подпунктов "а" - "в" настоящего пункта;
д) комбинированный лекарственный препарат, рассмотренный и одобренный Экспертным комитетом при невозможности выполнения подпунктов "а" - "б" настоящего пункта.
Экспертным комитетом выбор в качестве референтного комбинированного лекарственного препарата, содержащего известные действующие вещества и зарегистрированного как минимум в одном из государств-членов, при ограниченной программе его изучения, не позволяющей признать оригинальность, может быть сделан с учетом оценки длительности медицинского применения препарата и обоснованности создания фиксированной комбинации, при невозможности выполнения подпунктов "а" - "г" настоящего пункта.
При невозможности признать лекарственный препарат референтным по причине нерациональности комбинации, недоказанной эффективности и безопасности для разработки нового лекарственного препарата предлагается проведение программы доклинических и клинических исследований, соответствующей разработке новых комбинаций, в соответствии с актами органов Союза в сфере обращения лекарственных средств.".
3. В пункте 104 слова "государств - членов Союза и (или) Экспертный комитет по лекарственным средствам при Евразийской экономической комиссии" заменить словами "государств-членов и (или) Экспертный комитет".
4. Приложение N 4 изложить в следующей редакции:
"Приложение N 4
к Правилам проведения исследований
биоэквивалентности лекарственных
препаратов в рамках Евразийского
экономического союза
ТРЕБОВАНИЯ
К БИОВЕЙВЕРУ, ОСНОВАННОМУ НА БИОФАРМАЦЕВТИЧЕСКОЙ
СИСТЕМЕ КЛАССИФИКАЦИИ
I. Общие положения
1. Два лекарственных препарата, содержащих одинаковое действующее вещество (действующие вещества), считаются биоэквивалентными, если их биодоступность (скорость и степень абсорбции действующего вещества) после применения в одной и той же молярной дозе укладывается в заранее установленные приемлемые границы. Такие границы устанавливаются для обеспечения сопоставимого действия in vivo исследуемого лекарственного препарата (сопоставимости по его безопасности и эффективности с оригинальным (референтным) лекарственным препаратом). Для оценки скорости и степени абсорбции действующего вещества в исследованиях биоэквивалентности in vivo используются опорные фармакокинетические параметры AUC и Cmax.
2. Подход, основанный на применении биофармацевтической системы классификации (далее - БКС) при биовейвере, направлен на сокращение потребности в проведении исследований биоэквивалентности in vivo, (может служить суррогатным исследованием для оценки биоэквивалентности in vivo). Допускается не проводить исследования биоэквивалентности in vivo, если предположение об эквивалентности действия in vivo обосновано с помощью удовлетворительных данных in vitro. БКС разделяет действующие вещества на четыре класса:
класс I: хорошая растворимость, хорошая проникающая способность;
класс II: плохая растворимость, хорошая проникающая способность;
класс III: хорошая растворимость, плохая проникающая способность;
класс IV: плохая растворимость, плохая проникающая способность.
3. Настоящие Требования содержат указания по установлению класса БКС для действующего вещества и применения к нему биовейвера, основанного на БКС при замене исследований биоэквивалентности in vivo лекарственных препаратов. Принципы биовейвера, основанного на БКС, применимы также для целей исследования биоэквивалентности лекарственных форм лекарственных препаратов, не указанных в настоящих Требованиях, при условии, что указанные принципы научно обоснованы.
II. Критерии применения биовейвера, основанного на БКС
4. Биовейвер, основанный на БКС, используется для обоснования биоэквивалентности in vivo. Данная процедура применяется для проведения взаимного сравнения фармацевтической продукции, использованной в период, начиная с клинической разработки лекарственного препарата до выпуска его промышленных серий, оценки пострегистрационных изменений и заявления на регистрацию в качестве воспроизведенного лекарственного препарата.
5. Биовейвер, основанный на БКС, применяется только к твердым лекарственным формам для приема внутрь или суспензиям с немедленным высвобождением, предназначенным для доставки действующего вещества в системный кровоток. В отношении лекарственных препаратов, имеющих узкий терапевтический индекс, не допускается использование биовейвера, основанного на БКС, для замены исследований биоэквивалентности in vivo. Для комбинации фиксированных доз лекарственных препаратов применяется биовейвер, основанный на БКС, в случае, если все действующие вещества, содержащиеся в таком комбинированном лекарственном препарате, отвечают критериям, сформулированным в подразделах 2 и 3 раздела III настоящих Требований.
6. Биовейвер, основанный на БКС, применяется к лекарственным препаратам, если действующее вещество (вещества) имеет хорошую растворимость и хорошую проникающую способность (класс I по БКС) или хорошую растворимость и плохую проникающую способность (класс III по БКС).
7. Биовейвер, основанный на БКС, применяется в случае, если действующее вещество (вещества) исследуемого и референтного лекарственных препаратов идентично. Биовейвер, основанный на БКС, также применяется в случае, если исследуемый и референтный лекарственные препараты содержат разные соли при условии, что обе соли принадлежат классу I по БКС. Биовейвер, основанный на БКС, не применяется, если исследуемый лекарственный препарат содержит действующее вещество в виде другого простого эфира, сложного эфира, изомера, смеси изомеров, комплекса или иного производного, отличного от действующего вещества референтного лекарственного препарата, поскольку эти различия могут приводить к различиям в биодоступности, которые не могут быть получены в эксперименте, основанном на применении концепции БКС для биовейвера. К пролекарствам допускается применять биовейвер, основанный на БКС, если они абсорбируются в форме пролекарств.
III. Биофармацевтическая система классификации
действующих веществ
1. Растворимость
8. Действующее вещество классифицируется в качестве хорошо растворимого, если максимальная разовая терапевтическая доза (в соответствии с общей характеристикой референтного лекарственного препарата) полностью растворима в буферной среде объемом 250 мл или в меньшем объеме среды в диапазоне pH 1,2 - 6,8 при температуре 37 ![]() 1 °C. В случаях если максимальная разовая терапевтическая доза не отвечает этому критерию, но навеска действующего вещества, соответствующая наибольшей дозировке референтного лекарственного препарата, полностью растворима в вышеуказанных условиях, для обоснования биовейвера, основанного на БКС, в регистрационном досье лекарственного препарата должны быть представлены дополнительные данные.
1 °C. В случаях если максимальная разовая терапевтическая доза не отвечает этому критерию, но навеска действующего вещества, соответствующая наибольшей дозировке референтного лекарственного препарата, полностью растворима в вышеуказанных условиях, для обоснования биовейвера, основанного на БКС, в регистрационном досье лекарственного препарата должны быть представлены дополнительные данные.
9. Необходимо экспериментально установить растворимость действующего вещества в диапазоне pH 1,2 - 6,8 при температуре 37 ![]() 1 °C. Должны быть изучены как минимум три pH в этом диапазоне, включая буферные среды с pH 1,2, 4,5 и 6,8. Дополнительно должна быть изучена растворимость действующего вещества при pH с самой плохой его растворимостью, если это значение pH находится внутри указанного диапазона. Указанные исследования должны подтверждать, что растворимость действующего вещества поддерживается во временных рамках, соответствующих ожидаемой продолжительности абсорбции действующего вещества.
1 °C. Должны быть изучены как минимум три pH в этом диапазоне, включая буферные среды с pH 1,2, 4,5 и 6,8. Дополнительно должна быть изучена растворимость действующего вещества при pH с самой плохой его растворимостью, если это значение pH находится внутри указанного диапазона. Указанные исследования должны подтверждать, что растворимость действующего вещества поддерживается во временных рамках, соответствующих ожидаемой продолжительности абсорбции действующего вещества.
10. Растворимость необходимо оценивать с помощью метода, подобранного исходя из свойств действующего вещества.
11. Допускается проведение исследований равновесной растворимости с использованием методики встряхивания колбы или альтернативной методики, если это обосновано в регистрационном досье лекарственного препарата. Допускается использование небольших объемов буферной среды для оценки растворимости, если доступный для исследований аппарат позволяет сделать это. Для обеспечения измерения растворимости при установленном значении pH для каждого испытуемого раствора необходимо проводить измерение значения pH после добавления действующего вещества, а также в конце исследования равновесной растворимости. При необходимости значение pH корректируется. Эксперимент должен проводиться в течение периода времени, достаточного для того чтобы достичь равновесия.
12. В качестве альтернативного варианта исследований может быть проведено изучение растворимости, в котором максимальная разовая терапевтическая доза действующего вещества изучается в буферной среде объемом 250 мл или меньшая доза действующего вещества - в пропорционально меньшем объеме буферной среды.
13. Для определения класса по БКС действующего вещества должна быть использована наименьшая измеренная растворимость в диапазоне pH 1,2 - 6,8.
14. Для подтверждения полученного значения растворимости необходимо как минимум 3 повторных определения растворимости при каждом условии или pH с использованием соответствующих фармакопейных буферных сред и валидированного метода определения действующего вещества. В дополнение к экспериментальным данным в регистрационном досье лекарственного препарата допускается представление научных химико-технологических и фармацевтических данных, с целью получения доказательства и обоснования значении растворимости. При этом необходимо учитывать, что не все статьи научных рецензируемых журналов (изданий) содержат данные об исследованиях, необходимые для оценки качества этих исследований.
15. Необходимо подтверждение достаточной стабильности действующего вещества в буферной среде оценки растворимости. В случаях если действующее вещество нестабильно (его деградация составляет > 10% во время оценки растворимости), невозможно правильно определить растворимость и класс по БКС действующего вещества.
2. Проникающая способность
16. Оценка проникающей способности должна предпочтительно основываться на определении степени абсорбции действующего вещества, полученной из фармакокинетических исследований с участием человека (например, при изучении абсолютной биодоступности или материального баланса).
17. Если абсолютная биодоступность действующего вещества составляет ![]() 85%, можно сделать вывод о хорошей проникающей способности. Вывод о хорошей проникающей способности действующего вещества также можно сделать, если
85%, можно сделать вывод о хорошей проникающей способности. Вывод о хорошей проникающей способности действующего вещества также можно сделать, если ![]() 85% его введенной дозы выделяется в мочу в неизмененном виде (в виде исходного действующего вещества) или в виде суммы исходного действующего вещества, его окисленных (I фаза) и конъюгированных (II фаза) метаболитов. Для метаболитов, обнаруживаемых в фекалиях, также допускается учитывать только окисленные и конъюгированные метаболиты. Метаболиты, образующиеся вследствие восстановления или гидролиза, включать в общую оценку биодоступности не разрешается, если только нельзя продемонстрировать, что они не образуются до абсорбции (например, за счет влияния микроорганизмов в желудочно-кишечном тракте). Неизмененное действующее вещество в фекалиях нельзя включать в оценку степени абсорбции, если только соответствующие данные не обосновывают, что количество исходного действующего вещества в фекалиях, учитываемое в качестве абсорбированного действующего вещества, обусловлено его билиарной экскрецией, кишечной секрецией или образуется из нестабильного метаболита (например, глюкуронида, сульфата, N-оксида), который подвергается обратному преобразованию в исходное соединение под влиянием микроорганизмов.
85% его введенной дозы выделяется в мочу в неизмененном виде (в виде исходного действующего вещества) или в виде суммы исходного действующего вещества, его окисленных (I фаза) и конъюгированных (II фаза) метаболитов. Для метаболитов, обнаруживаемых в фекалиях, также допускается учитывать только окисленные и конъюгированные метаболиты. Метаболиты, образующиеся вследствие восстановления или гидролиза, включать в общую оценку биодоступности не разрешается, если только нельзя продемонстрировать, что они не образуются до абсорбции (например, за счет влияния микроорганизмов в желудочно-кишечном тракте). Неизмененное действующее вещество в фекалиях нельзя включать в оценку степени абсорбции, если только соответствующие данные не обосновывают, что количество исходного действующего вещества в фекалиях, учитываемое в качестве абсорбированного действующего вещества, обусловлено его билиарной экскрецией, кишечной секрецией или образуется из нестабильного метаболита (например, глюкуронида, сульфата, N-оксида), который подвергается обратному преобразованию в исходное соединение под влиянием микроорганизмов.
18. Допускается представлять данные in vivo, полученные в результате проведения исследований с участием человека, взятые из опубликованной научной медицинской литературы (например, знания свойств лекарственного препарата и исследования биодоступности). При этом необходимо учитывать, что не все статьи научных рецензируемых журналов (изданий) содержат необходимые данные об исследованиях, чтобы оценить качество этих исследований.
19. Анализ проникающей способности действующего вещества также проводится с помощью валидированных и стандартизированных методов исследований in vitro с использованием линии клеток Caco-2 (в соответствии с Методикой согласно приложению N 1). Результаты изучения проникающей способности действующего вещества на линии клеток Caco-2 необходимо проанализировать с учетом имеющихся данных о фармакокинетике действующего вещества у человека. Если заключение о хорошей проникающей способности действующего вещества выносится на основе изучения in vitro клеточной системы, необходимо доказать наличие проникающей способности действующего вещества, не зависящей от его активного транспорта, в соответствии с приложением N 1 к настоящим Требованиям.
20. Если хорошая проникающая способность не установлена, действующее вещество признается обладающим плохой проникающей способностью для его классификации по БКС.
3. Стабильность действующего вещества
в желудочно-кишечном тракте
21. В случае если для подтверждения хорошей проникающей способности действующего вещества используются исследования материального баланса, необходимо представление дополнительных данных, содержащих сведения об оценке стабильности действующего вещества в желудочно-кишечном тракте, за исключением случаев, если ![]() 85% введенной дозы действующего вещества выводится с мочой в виде неизмененного действующего вещества. Подтверждение стабильности действующего вещества в желудочно-кишечном тракте является обязательным, если для обоснования его хорошей проникающей способности используются исследования in vitro на линии клеток Caco-2. Стабильность действующего вещества в желудочно-кишечном тракте необходимо документировать с использованием фармакопейных сред или сред, имитирующих желудочный и кишечный соки. При наличии надлежащего обоснования в регистрационном досье лекарственного препарата допускается использование других релевантных методов оценки стабильности действующего вещества. Растворы активной фармацевтической субстанции необходимо инкубировать при температуре 37,0
85% введенной дозы действующего вещества выводится с мочой в виде неизмененного действующего вещества. Подтверждение стабильности действующего вещества в желудочно-кишечном тракте является обязательным, если для обоснования его хорошей проникающей способности используются исследования in vitro на линии клеток Caco-2. Стабильность действующего вещества в желудочно-кишечном тракте необходимо документировать с использованием фармакопейных сред или сред, имитирующих желудочный и кишечный соки. При наличии надлежащего обоснования в регистрационном досье лекарственного препарата допускается использование других релевантных методов оценки стабильности действующего вещества. Растворы активной фармацевтической субстанции необходимо инкубировать при температуре 37,0 ![]() 0,5 °C в течение срока, соответствующего времени контакта in vivo действующего вещества с этими соками, а именно: 1 час - в желудочном соке и 3 часа в - кишечном соке. Концентрации действующего вещества в растворе затем определяется с использованием валидированного метода. Значительная деградация действующего вещества в объеме > 10% не позволяет классифицировать его в соответствии с БКС в качестве хорошо проникающего действующего вещества.
0,5 °C в течение срока, соответствующего времени контакта in vivo действующего вещества с этими соками, а именно: 1 час - в желудочном соке и 3 часа в - кишечном соке. Концентрации действующего вещества в растворе затем определяется с использованием валидированного метода. Значительная деградация действующего вещества в объеме > 10% не позволяет классифицировать его в соответствии с БКС в качестве хорошо проникающего действующего вещества.
IV. Пригодность лекарственного препарата для биовейвера,
основанного на БКС
22. Лекарственный препарат является пригодным для биовейвера, основанного на БКС, при выполнении всех следующих условий:
действующее вещество (вещества) исследуемого лекарственного препарата соответствует классам I и III по БКС по критериям растворимости и проникающей способности;
исследуемый и референтный лекарственные препараты являются лекарственными формами для приема внутрь с немедленным высвобождением и системным действием;
исследуемый лекарственный препарат имеет ту же лекарственную форму и дозировку, что и референтный препарат.
23. В случаях если максимальная разовая терапевтическая доза лекарственного препарата не удовлетворяет критерию хорошей растворимости, но наибольшая дозировка референтного препарата растворима в требуемых условиях, применение биовейвера, основанного на БКС, необходимо обосновать с помощью демонстрации дозопропорциональной фармакокинетики исследуемого лекарственного препарата (AUC и Cmax) в диапазоне доз, включающем максимальную разовую терапевтическую дозу.
24. Лекарственные препараты с буккальной или сублингвальной абсорбцией являются не пригодными для применения биовейвера, основанного на БКС. Подход, основанный на использовании биофармацевтической системы классификации, применяется при биовейвере, только в случае если лекарственный препарат принимают запивая водой. Если для лекарственного препарата также допускается прием без запивания водой (например, лекарственные препараты, диспергируемые в полости рта), необходимо проведение исследования биоэквивалентности in vivo, в котором лекарственный препарат будет применяться без запивания водой.
25. Чтобы лекарственный препарат мог претендовать на применение биовейвера, основанного на БКС, должны выполняться критерии, предъявляемые к составу (вспомогательным веществам) и характеристикам растворения in vitro лекарственного препарата, указанные в подразделах 1 и 2 настоящего раздела настоящих Требований.
1. Вспомогательные вещества
26. Необходимо добиваться того, чтобы состав исследуемого лекарственного препарата полностью воспроизводил состав референтного лекарственного препарата. В случае наличия различий между вспомогательными веществами исследуемого и референтного лекарственных препаратов, их необходимо оценить с точки зрения потенциального влияния таких различий на абсорбцию действующего вещества in vivo. Такая оценка включает в себя анализ свойств действующего вещества и влияния вспомогательных веществ на его абсорбцию. В целях применения биовейвера, основанного на БКС, спонсор обязан обосновать, почему предлагаемые им различия во вспомогательных веществах лекарственных препаратов не будут влиять на профиль абсорбции рассматриваемого действующего вещества (его скорость и степень абсорбции), используя физико-химические исследования и расчеты, а также риск-ориентированный подход.
Дерево решений для проведения подобной оценки представлено на рисунках 1 и 2 приложения N 2 к настоящим Требованиям.
27. Заявителю необходимо проанализировать возможное влияние вспомогательных веществ на следующие аспекты абсорбции действующего вещества in vivo:
растворимость самого действующего вещества;
влияние вспомогательных веществ на желудочно-кишечную моторику;
время транзита действующего вещества и его кишечную проникающую способность, включая механизмы переноса через мембраны клеток.
28. Вспомогательные вещества, способные влиять на абсорбцию, включают в себя альдолы (спиртосахара, например, маннитол, сорбитол), а также поверхностно активные вещества (например, натрия додецил сульфат).
29. Необходимо оценить возможный риск влияния вспомогательного вещества на абсорбцию действующего вещества, путем проведения физико-химических расчетов с учетом:
а) количества используемого в единице лекарственной формы вспомогательного вещества;
б) механизма, с помощью которого вспомогательное вещество может повлиять на абсорбцию действующего вещества;
в) абсорбционных свойств (скорость, степень и механизм абсорбции) действующего вещества.
30. Во время фармацевтической разработки лекарственного препарата необходимо учитывать в составе исследуемого и референтного препаратов количество вспомогательных веществ, способных влиять на абсорбцию действующего вещества, с целью обеспечения минимального изменения состава и содержания вспомогательных веществ в исследуемом лекарственном препарате. Допускается не учитывать незначительное количество вспомогательных веществ, включаемых в оболочку таблетки, или количество вспомогательных веществ ниже их установленного (доказанного) порога влияния.
31. Действующие вещества класса I по БКС представляют собой группу соединений низкого риска с точки зрения способности вспомогательных веществ в их составе влиять на абсорбцию по сравнению с другими классами веществ по БКС поскольку:
хорошо абсорбируются;
имеют не ограниченную ни растворимостью, ни проникающей способностью абсорбцию.
32. Оценка влияния вспомогательных веществ в отношении лекарственных препаратов, действующие вещества которых относятся к классу I по БКС, должна включать в себя анализ потенциальных изменений скорости или степени абсорбции. Например, если известно, что действующее вещество имеет хорошую проникающую способность благодаря активному захвату, невозможность применения биовейвера, основанного на БКС, будет связана с наличием в составе лекарственного препарата вспомогательных веществ, способных ингибировать захватывающие переносчики. В отношении действующих веществ класса I по БКС, обладающих медленной абсорбцией, необходимо также учитывать способность входящих в состав лекарственного препарата вспомогательных веществ повышать его абсорбцию.
33. В отношении лекарственных препаратов, действующие вещества которых относятся к классу I по БКС, допустимы качественные и количественные различия во вспомогательных веществах исследуемого и референтного лекарственного препарата, за исключением вспомогательных веществ, способных влиять на абсорбцию действующего вещества, которые должны совпадать качественно и быть схожими по количественному составу (отличаться в пределах ![]() 10% от количественного содержания вспомогательного вещества в референтном препарате). Суммарная разница в содержании всех вспомогательных веществ, способных влиять на абсорбцию, должна быть в пределах
10% от количественного содержания вспомогательного вещества в референтном препарате). Суммарная разница в содержании всех вспомогательных веществ, способных влиять на абсорбцию, должна быть в пределах ![]() 10%.
10%.
34. Поведение в организме действующих веществ класса III по БКС в большей степени зависит от влияния вспомогательных веществ. Эти действующие вещества не обладают хорошей проникающей способностью и могут иметь абсорбцию в специфическом отделе желудочно-кишечного тракта, поэтому существует много механизмов, с помощью которых вспомогательные вещества способны повлиять на их абсорбцию, по сравнению с действующими веществами класса I по БКС. В отношении действующих веществ класса III по БКС все вспомогательные вещества должны быть одинаковыми по качественному составу и схожими по количественному составу (за исключением вспомогательных веществ пленочной оболочки или оболочки капсулы) с составом референтного лекарственного препарата. Вспомогательные вещества, способные влиять на абсорбцию действующих веществ, должны быть идентичными по качественному составу и схожими по количественному составу с составом референтного лекарственного препарата (отличаться в пределах ![]() 10% от содержания вспомогательного вещества в референтном препарате, а суммарная разница в содержании всех вспомогательных веществ, способных влиять на абсорбцию, должна быть в пределах
10% от содержания вспомогательного вещества в референтном препарате, а суммарная разница в содержании всех вспомогательных веществ, способных влиять на абсорбцию, должна быть в пределах ![]() 10%). Необходимо придерживаться параметров, подтверждающих сходство по количественному составу вспомогательных веществ исследуемого и референтного лекарственных препаратов, приведенных в таблице. Примеры допустимых различий во вспомогательных веществах приведены в приложении N 2 к настоящим Требованиям. Различия в красителях и ароматизаторах допустимы, если их содержание в составе лекарственного препарата незначительное.
10%). Необходимо придерживаться параметров, подтверждающих сходство по количественному составу вспомогательных веществ исследуемого и референтного лекарственных препаратов, приведенных в таблице. Примеры допустимых различий во вспомогательных веществах приведены в приложении N 2 к настоящим Требованиям. Различия в красителях и ароматизаторах допустимы, если их содержание в составе лекарственного препарата незначительное.
35. В ряде случаев (например, при проблемах с определением массы пленочной оболочки референтного препарата) применение параметров, приведенных в таблице, бывает затруднено. В таблице приводятся ориентировочные параметры, которые необходимо учитывать в процессе фармацевтической разработки. Отклонения от указанных показателей требуют соответствующего обоснования в регистрационном досье лекарственного препарата исходя из принципов, указанных в пунктах 26 - 34 настоящих Требований.
Таблица
Параметры, подтверждающие сходство по количественному
составу вспомогательных веществ исследуемого и референтного
лекарственных препаратов, содержащих действующее
вещество класса III по БКС
Вид вспомогательного вещества
Отклонение от массы, не более
Вспомогательные вещества, влияющие на абсорбцию:
индивидуальное вещество
10% <1>
все вещества суммарно
10% <1>
Прочие вспомогательные вещества:
наполнитель
10% <2>
Дезинтегранты:
крахмал
6% <2>
иной
2% <2>
Связующее вещество
1% <2>
Смазывающие вещества (лубриканты):
стеараты
0,5% <2>
другие
2% <2>
Скользящие вещества:
тальк
2% <2>
иные
0,2% <2>
Изменения всех вспомогательных веществ суммарно, включая вещества, влияющие на абсорбцию
10% <2>
Примечание:
<1> Отклонение от массы вспомогательного вещества в референтном препарате.
<2> Отклонение от массы ядра лекарственной формы референтного препарата. Масса ядра не включает массу пленочной оболочки таблетки или оболочку капсулы.
36. Биовейвер, основанный на БКС, применим к комбинациям фиксированных доз действующих веществ при одинаковой лекарственной форме и дозировке у исследуемого и референтного лекарственных препаратов. Указанные лекарственные препараты, содержащие действующие вещества класса I, должны отвечать критериям в отношении вспомогательных веществ для действующего вещества класса I по БКС. Препараты комбинаций фиксированных доз, содержащие действующие вещества класса III по БКС или действующие вещества класса I и класса III по БКС, должны отвечать критериям в отношении вспомогательных веществ для действующего вещества класса III по БКС.
2. Тест сравнительной кинетики растворения in vitro
для лекарственного препарата
37. При применении биовейвера, основанного на БКС, необходимо провести тест сравнительной кинетики растворения in vitro с использованием одной промышленной или опытно-промышленной серии лекарственного препарата, соответствующей заявляемому на регистрацию процессу ее производства, включающий в себя сравнение с референтным лекарственным препаратом.
38. Исследуемый лекарственный препарат необходимо брать из серии, размер которой составляет не менее 1/10 размера промышленной серии или 100 000 единиц (в зависимости от того, что больше), если заявителем (производителем) не обосновано иное. При проведении теста сравнительной кинетики растворения in vitro для выбора лекарственного препарата исходя из фазы его клинической разработки допускается использование опытно-промышленной серии лекарственного препарата меньших размеров при обосновании. В тестах сравнительной кинетики растворения in vitro необходимо использовать валидированный аналитический метод (аналитические методы), а также прибор, соответствующий Фармакопее Евразийского экономического союза, утвержденной Решением Коллегии Евразийской экономической комиссии от 11 августа 2020 г. N 100.
39. В целях определения профиля растворения лекарственных препаратов в тесте сравнительной кинетики in vitro растворения необходимо выполнение следующих условий:
прибор: Прибор 2 (лопастная мешалка) или Прибор 1 (вращающаяся корзинка);
объем среды растворения: 900 мл или меньше (предпочтительно использовать объем, выбранный для среды контроля качества);
температура среды растворения: 37,0 ![]() 1,0 °C;
1,0 °C;
скорость перемешивания: Прибор 2 (лопастная мешалка) - 50 оборотов в минуту, Прибор 1 (вращающаяся корзинка) - 100 оборотов в минуту;
при каждом экспериментальном определении профиля растворения необходимо использование не менее 12 дозовых единиц референтного и исследуемого лекарственных препаратов;
три буферных среды: pH 1,2, pH 4,5 и pH 6,8. Необходимо использовать фармакопейные буферы. При pH в котором наблюдается минимальная растворимость лекарственного препарата (если эта среда отличается от вышеуказанных буферов), необходимо предусмотреть проведение дополнительного изучения;
не допускается использование органических растворителей и добавление поверхностно активных веществ в среду растворения;
во время отбора проб необходимо обеспечить их фильтрование, за исключением случаев, когда используются методы обнаружения действующего вещества in situ;
в отношении желатиновых капсул или таблеток с желатиновой оболочкой, если в их составе установлено наличие перекрестных сшивок молекул желатина, допускается использование ферментов при наличии обоснования.
40. Если в Приборе 2 (лопастная мешалка) на скорости 50 оборотов в минуту наблюдается высокая вариабельность высвобождения действующего вещества или конусообразование как для референтного, так и исследуемого лекарственных препаратов, допускается использование Прибора 1 (вращающаяся корзинка) на скорости 100 оборотов в минуту. Кроме того, допускается использование альтернативных методов (например, использование грузил (спикеров) или других должным образом обоснованных подходов) для устранения конусообразования и других проблем. В отчете о проведенном исследовании необходимо представить все результаты экспериментов.
41. Для сравнения профилей растворения там, где применимо, необходимо оценить фактор подобия f2 но следующей формуле:
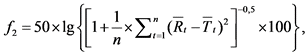
где:
f2 - фактор подобия;
n - число временных точек;
R(t) - средняя доля (в процентах) растворившегося действующего вещества референтного лекарственного препарата в момент времени t после начала исследования;
T(t) - средняя доля (в процентах) растворившегося действующего вещества исследуемого лекарственного препарата в момент времени t после начала исследования.
42. Расчет фактора подобия применяется при выполнении всех следующих условий:
проведена оценка растворения по меньшей мере в трех временных точках (не считая нулевой точки отбора);
выбранные временные точки отбора проб для двух сравниваемых лекарственных препаратов должны быть одинаковыми;
для каждой временной точки рассчитано среднее значение индивидуальных показателей растворения действующего вещества каждого из сравниваемых лекарственных препаратов;
наличие для любого из препаратов не более одной временной точки со средним значением растворения действующего вещества ![]() 85% от его номинального содержания в лекарственном препарате;
85% от его номинального содержания в лекарственном препарате;
коэффициент вариации для средних значений растворения действующего вещества лекарственного препарата не должен превышать 20% в ранние временные точки (до 10 минут) и 10% в остальные временные точки.
43. Два профиля растворения действующих веществ сравниваемых лекарственных препаратов считаются подобными, если значение f2 ![]() 50. Если как у исследуемого, так и у референтного лекарственных препаратов установлено, что
50. Если как у исследуемого, так и у референтного лекарственных препаратов установлено, что ![]() 85% заявленного количества действующего вещества растворяется за 15 минут, сравнение с помощью оценки фактора подобия f2 не требуется, а профили растворения признаются подобными. Если коэффициент вариации для средних значений растворения действующего вещества превышает значения, указанные в пункте 42 настоящих Требований, расчет фактора подобия f2 признается неправильным, и сделать вывод о подобии (схожести) профилей растворения действующего вещества у сравниваемых лекарственных препаратов невозможно.
85% заявленного количества действующего вещества растворяется за 15 минут, сравнение с помощью оценки фактора подобия f2 не требуется, а профили растворения признаются подобными. Если коэффициент вариации для средних значений растворения действующего вещества превышает значения, указанные в пункте 42 настоящих Требований, расчет фактора подобия f2 признается неправильным, и сделать вывод о подобии (схожести) профилей растворения действующего вещества у сравниваемых лекарственных препаратов невозможно.
44. Для применения биовейвера, основанного на БКС, в отношении действующих веществ класса I по БКС исследуемый и референтный лекарственные препараты должны проявлять:
либо очень быстрые (![]() 85% - для среднего количества вещества, перешедшего в раствор за
85% - для среднего количества вещества, перешедшего в раствор за ![]() 15 минут) характеристики профиля растворения in vitro;
15 минут) характеристики профиля растворения in vitro;
либо быстрые (![]() 85% - для среднего количества вещества, перешедшего в раствор за
85% - для среднего количества вещества, перешедшего в раствор за ![]() 30 минут) и схожие (подобные) характеристики профиля растворения in vitro (например, на основании сравнения по фактору подобия f2) во всех условиях растворения.
30 минут) и схожие (подобные) характеристики профиля растворения in vitro (например, на основании сравнения по фактору подобия f2) во всех условиях растворения.
В случае если один из препаратов (исследуемый или референтный) имеет быстрое растворение, а другой препарат - очень быстрое растворение, должно быть продемонстрировано подобие профилей на основании сравнения по фактору f2.
45. Для применения биовейвера, основанного на БКС, в отношении лекарственных препаратов, содержащих действующие вещества класса III по БКС исследуемый и референтный лекарственные препараты должны проявлять очень быстрые (![]() 85% - для среднего количества вещества, перешедшего в раствор за
85% - для среднего количества вещества, перешедшего в раствор за ![]() 15 минут) и схожие (подобные) характеристики профиля растворения in vitro во всех условиях растворения.
15 минут) и схожие (подобные) характеристики профиля растворения in vitro во всех условиях растворения.
46. Для применения биовейвера, основанного на БКС, в отношении лекарственных препаратов с фиксированной комбинацией доз должны быть подобны профили растворения всех действующих веществ в составе рассматриваемых лекарственных препаратов с фиксированной комбинацией доз. Оценка подобия профилей растворения препаратов с фиксированной комбинацией доз зависит от их состава:
препараты, содержащие исключительно действующие вещества класса I по БКС, должны отвечать критериям подобия профилей растворения для каждого действующего вещества класса I по БКС;
препараты, содержащие исключительно действующие вещества класса III по БКС, должны отвечать критериям подобия профилей растворения для каждого действующего вещества класса III по БКС;
препараты, содержащие комбинацию действующих веществ класса I и III по БКС, должны отвечать критериям подобия профилей растворения, соответствующего класса БКС для каждого компонента комбинации.
47. В отношении лекарственных препаратов с более чем одной дозировкой подход к применению биовейвера, основанного на БКС, должен применяться к каждой дозировке лекарственного препарата и включает в себя сравнение профилей растворения исследуемого и референтного лекарственных препаратов для каждой дозировки.
Дополнения к условиям применения требований к биовейверу, основанному на БКС, приведены в приложении N 3.
V. Документы, необходимые для представления в составе
регистрационного досье, при использовании биовейвера,
основанного на БКС
48. Спонсор обязан представить сведения о критичных показателях качества изучаемого действующего вещества (веществ) и лекарственного препарата, а также информацию о референтном препарате, включающую в себя в том числе:
вид полиморфной формы активной фармацевтической субстанции;
энантиомерную чистоту активной фармацевтической субстанции;
любые сведения о проблемах биодоступности или биоэквивалентности действующего вещества (веществ) или лекарственного препарата, включая медицинские научные данные и данные проведенных собственных исследований.
Необходимо представить все протоколы и отчеты об исследованиях. Сведения о валидации аналитических методов должны соответствовать:
требованиям, изложенным в приложении N 6 к Правилам проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза, утвержденным Решением Совета Евразийской экономической комиссии от 3 ноября 2016 г. N 85 - для биоаналитических методик;
требованиям Руководства по валидации аналитических методик проведения испытаний лекарственных средств, утвержденного Решением Коллегии Евразийской экономической комиссии от 17 июля 2018 г. N 113 - для аналитических методик.
49. Отчет должен включать в себя описание всех вспомогательных веществ, входящих в состав исследуемого и референтного лекарственных препаратов, с приведением их качественных и если применимо количественных различий.
50. В отчете необходимо представить полное описание использованных аналитических методик, включая их валидацию и квалификацию аналитического оборудования. Дополнительно необходимо представить полное описание всех сред растворения лекарственных препаратов, а также сведения об исследуемой и референтной сериях лекарственных препаратов (единичная доза (дозировка), использованная в тесте сравнительной кинетики растворения, протоколы анализа серии), номер серии, дату производства и размер серии (если они известны), дату истечения срока годности (срока хранения). Отчет об определении растворимости активной фармацевтической субстанции и проведении теста сравнительной кинетики растворения лекарственного препарата должен включать в себя подробное описание экспериментальных условий и аналитических методик, включая такие сведения об условиях растворения, как:
использованный фармакопейный прибор;
методики деаэрации;
способы фильтрации в процессе отбора проб;
объем отбираемых проб;
другие параметры, позволяющие описать процесс получения экспериментальных данных.
51. Дополнительно в отчете необходимо представить сведения с полным описанием использованных методов определения проникающей способности действующего вещества на линии клеток Caco-2, если она применялась (в соответствии с положениями, указанными в приложении N 1 к настоящим Требованиям).
52. Отчет, указанный в пунктах 49 и 50 настоящих Требований, должен содержать табличные и графические данные, соответствующие индивидуальным и средним результатам, а также обобщающие статистические данные.
Приложение N 1
к Требованиям к биовейверу,
основанному на биофармацевтической
системе классификации
МЕТОДИКА
АНАЛИЗА ПРОНИКАЮЩЕЙ СПОСОБНОСТИ ДЕЙСТВУЮЩЕГО ВЕЩЕСТВА
С ИСПОЛЬЗОВАНИЕМ ЛИНИИ КЛЕТОК CACO-2
1. Общие указания
В целях проведения оценки кишечной абсорбции действующего вещества у человека допускается проведение анализа проникающей способности этого действующего вещества с использованием культивируемых монослоев эпителиальных клеток Caco-2, получаемых из клеточной линии аденокарциномы толстой кишки человека. Клетки Caco-2 подвергаются спонтанной морфологической и биохимической дифференцировке в энтероциты и проявляют клеточную полярность, имея апикальную щеточную каемку, плотные межклеточные сочленения и несколько активных транспортеров, идентичных активным транспортерам в клетках тонкой кишки человека. Ввиду низкого или нулевого потенциала экспрессии эффлюкс-транспортеров (например, P-gp, PCRP, MRP2) и захватывающих транспортеров (например, PepT1, OATP2B1, MCT1) использование результатов анализа на линии клетках Caco-2 в качестве единственных данных для обоснования хорошей проникающей способности действующего вещества для целей их классификации по биофармацевтической системе классификации (далее - БКС) применимо только к действующим веществам, переносимым путем пассивной диффузии.
2. Валидация методики анализа проникающей способности
Пригодность анализов на линии клеток Caco-2 для определения проникающей способности действующего вещества по БКС подтверждается с помощью установления ранговой (порядковой) зависимости между экспериментальными значениями проникающей способности и степенью абсорбции действующего вещества у человека с использованием модельных действующих веществ с нулевой, плохой (< 50%), умеренной (50 - 84%) и хорошей (![]() 85%) проникающей способностью. Необходимо использовать достаточное количество модельных действующих веществ для валидации методики анализа с целью установления проникающей способности (по меньшей мере 5 действующих веществ на каждую степень проникающей способности - хорошая, умеренная и плохая), а также маркер характеризующий нулевую проникающую способность. Возможные примеры модельных действующих веществ приведены в таблице 2. Чтобы получить надежную оценку проникающей способности действующего вещества, необходимо использовать достаточное количество (по меньше мере 3) повторностей анализа на линии клеток Caco-2. Установленная ранговая (порядковая) зависимость должна позволять дифференцировать действующие вещества с плохой, умеренной и хорошей проникающей способностью.
85%) проникающей способностью. Необходимо использовать достаточное количество модельных действующих веществ для валидации методики анализа с целью установления проникающей способности (по меньшей мере 5 действующих веществ на каждую степень проникающей способности - хорошая, умеренная и плохая), а также маркер характеризующий нулевую проникающую способность. Возможные примеры модельных действующих веществ приведены в таблице 2. Чтобы получить надежную оценку проникающей способности действующего вещества, необходимо использовать достаточное количество (по меньше мере 3) повторностей анализа на линии клеток Caco-2. Установленная ранговая (порядковая) зависимость должна позволять дифференцировать действующие вещества с плохой, умеренной и хорошей проникающей способностью.
Должна быть подтверждена сохранность монослоя линии клеток Caco-2 до начала эксперимента и после его завершения при помощи сравнения показателей трансэпителиального электрического сопротивления и (или) других подходящих показателей.
Сохранность монослоя линии клеток Caco-2 должна быть продемонстрирована с помощью действующих веществ с доказанной нулевой проникающей способностью, перечисленных в таблице.
Описание процедуры валидации методики анализа должно включать в себя:
перечень модельных действующих веществ вместе с данными о степени их абсорбции у человека (среднее, стандартное отклонение, коэффициент вариации), использованных для установления пригодности метода;
значения проникающей способности каждого модельного действующего вещества (среднее, стандартное отклонение, коэффициент вариации);
указание класса проникающей способности каждого модельного действующего вещества;
график зависимости степени абсорбции действующего вещества от его проникающей способности (с представлением на графике значений в виде "среднее +/- стандартное отклонение" или 95% доверительного интервала);
указание границы хорошей проникающей способности и выбранного модельного действующего вещества для определения хорошей проникающей способности, использованных при определении класса по БКС исследуемого действующего вещества.
Дополнительно в отчете необходимо представить:
описание методики исследования;
использованные концентрации действующего вещества в донорской жидкости;
описание аналитической методики;
уравнение, использованное для расчета проникающей способности действующего вещества;
сведения об эффлюкс-потенциале линии клеток Caco-2 (например, в виде данных двунаправленного транспорта для известного действующего вещества).
3. Методика анализа проникающей способности
В ходе эксперимента должна быть установлена пассивная диффузия исследуемого действующего вещества. Наличие пассивной диффузии действующего вещества подтверждается на подходящей тест-системе, клетки которой экспрессируют известные эффлюкс-транспортеры. Одним из способов подтверждения пассивной диффузии является установление отсутствия зависимости измеренной проникающей способности in vitro действующего вещества и его исходной концентрации (например, отсутствие такой зависимости для дозировок, которые составляют 0,01, 0,10 и 1,00 от наибольшей дозировки действующего вещества, растворенной в 250 мл среды). Также пассивная диффузия может быть подтверждена путем оценки направления переноса действующего вещества (эффлюкс-отношения, то есть отношения кажущейся проникающей способности действующего вещества (Ркаж) между базолатерально-апикальным направлением клетки и апикально-базолатеральным направлением клетки), которое должно составлять < 2 для выбранных концентраций действующего вещества. Эффлюкс-отношение рассчитывается по следующей формуле:

где:
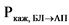 - кажущаяся проникающая способность действующего вещества в базолатерально-апикальном направлении;
- кажущаяся проникающая способность действующего вещества в базолатерально-апикальном направлении;
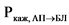 - кажущаяся проникающая способность действующего вещества в апикально-базолатеральном направлении.
- кажущаяся проникающая способность действующего вещества в апикально-базолатеральном направлении.
Функциональную экспрессию эффлюкс-транспортеров необходимо проверять с использованием исследований двунаправленного транспорта, подтверждающих асимметричную проникающую способность выбранных субстратов для эффлюкс-транспортеров (например, дигоксина, винбластина, родамина 123) используемых в ненасыщающих транспортеры концентрациях.
Концентрации исследуемого действующего вещества, используемые в исследованиях проникающей способности, должны быть обоснованы заявителем. В валидированной методике анализов проникающей способности действующего вещества на линии клеток Caco-2, необходимо использовать условия, установленные во время валидации методики, и включить модельные действующие вещества с умеренной и хорошей проникающей способностью в донорскую жидкость вместе с исследуемым действующим веществом в качестве внутренних стандартов для подтверждения постоянства методики указанного анализа. Выбор внутренних стандартов должен основываться на их совместимости с исследуемым действующим веществом, то есть они не должны проявлять какие-либо существенные физические, химические и опосредованные переносом взаимодействия с исследуемым действующим веществом. Проникающая способность внутренних стандартов может быть определена после оценки исследуемого действующего вещества в тех же монослоях линии клеток Caco-2 или монослоях клеток на том же планшете, если невозможно включить внутренние стандарты в ту же клеточную культуру, в которой оценивалась проникающая способность исследуемого действующего вещества. Полученные в серии различных испытаний, а также при валидации методики анализа значения проникающей способности внутренних стандартов должны коррелировать (быть близкими) между собой. В отчете должны быть указаны критерии приемлемости для внутренних стандартов и модельного субстрата для эффлюкс-транспортера. Необходимо указать среднее высвобождение действующего вещества и внутренних стандартов в конце испытания. При высвобождении действующего вещества < 80% должна быть выполнена оценка материального баланса, включая измерение остаточного количества действующего вещества в монослое линии клеток Caco-2 и в буферной среде которая находится в приборе для испытаний.
Оценку проникающей способности исследуемого действующего вещества для его классификации по БКС можно упростить в случае выбора внутреннего стандарта с хорошей проникающей способностью, значение которой находится вблизи установленной границы между умеренной и хорошей проникающей способностями. В этом случае исследуемое действующее вещество считается обладающим хорошей проникающей способностью, если значение его проникающей способности равно или превышает значение проникающей способности для выбранного внутреннего стандарта с хорошей проникающей способностью.
Сведения, необходимые для обоснования хорошей проникающей способности исследуемого действующего вещества (среднее значение проникающей способности, его стандартное отклонение, коэффициент вариации) должны включать в себя:
данные о проникающей способности исследуемого действующего вещества;
данные о проникающей способности использованных внутренних стандартов;
данные о желудочно-кишечной стабильности действующего вещества in vitro;
данные, обосновывающие механизм пассивной диффузии действующего вещества.
Таблица
Примеры модельных действующих веществ для валидации методики
анализа проникающей способности действующего вещества
с использованием линии клеток Caco-2
Класс проникающей способности или вид субстрата
Действующее вещество
Хорошая проникающая способность
(fa > 85%)
Антипирин
Кофеин
Кетопрофен
Напроксен
Теофиллин
Метопролол
Пропранолол
Карбамазепин
Фенитоин
Дизопирамид
Миноксидил
Умеренная проникающая способность
(fa = 50 - 84%)
Хлорамфеникол
Креатинин
Тербуталин
Гидрохоротиазид
Эналаприл
Фуросемид
Метформин
Амилорид
Атенолол
Ранитидин
Плохая проникающая способность
(fa < 50%)
Фамотидин
Надолол
Сульпирид
Лизиноприл
Ацикловир
Фоскарнет
Маннитол
Хлоротиазид
Полиэтиленгликоль 400
Эналаприлат
Нулевая проникающая способность
ФИТЦ-декстран
Полиэтиленгликоль 4000
Люцифер желтый
Инулин
Лактулоза
Модельные субстраты для эффлюкс-транспортеров
Дигоксин
Паклитаксел
Хинидин
Винбластин
Приложение N 2
к Требованиям к биовейверу,
основанному на биофармацевтической
системе классификации
АЛГОРИТМЫ И ПРИМЕРЫ
ОЦЕНКИ РАЗЛИЧИЙ В СОСТАВЕ ВСПОМОГАТЕЛЬНЫХ ВЕЩЕСТВ
В ИССЛЕДУЕМОМ И РЕФЕРЕНТНОМ ЛЕКАРСТВЕННЫХ ПРЕПАРАТАХ
1. Алгоритмы оценки различий в составе вспомогательных веществ в исследуемом и референтном лекарственных препаратах
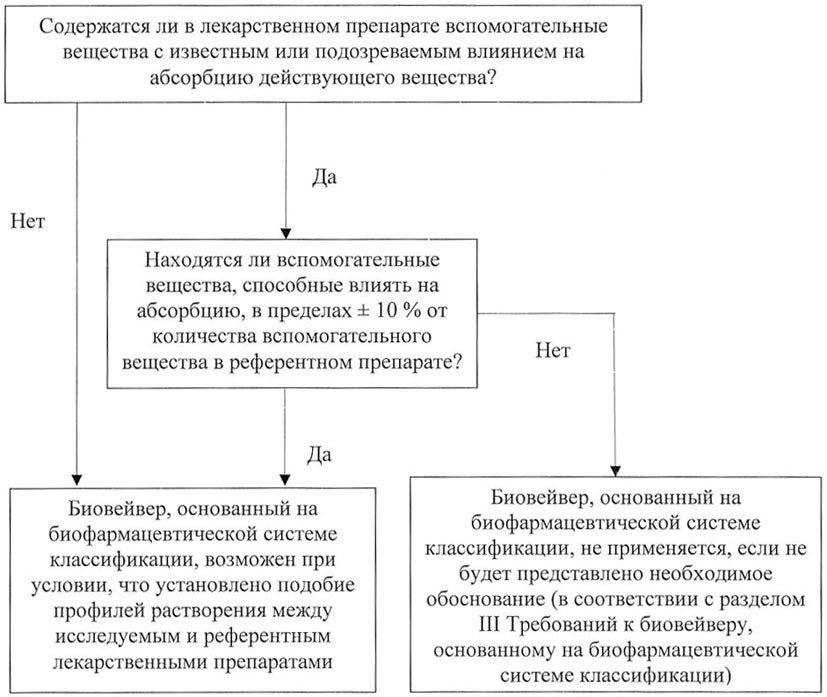
Рисунок 1. Алгоритм оценки лекарственного препарата,
содержащего действующие вещества класса I
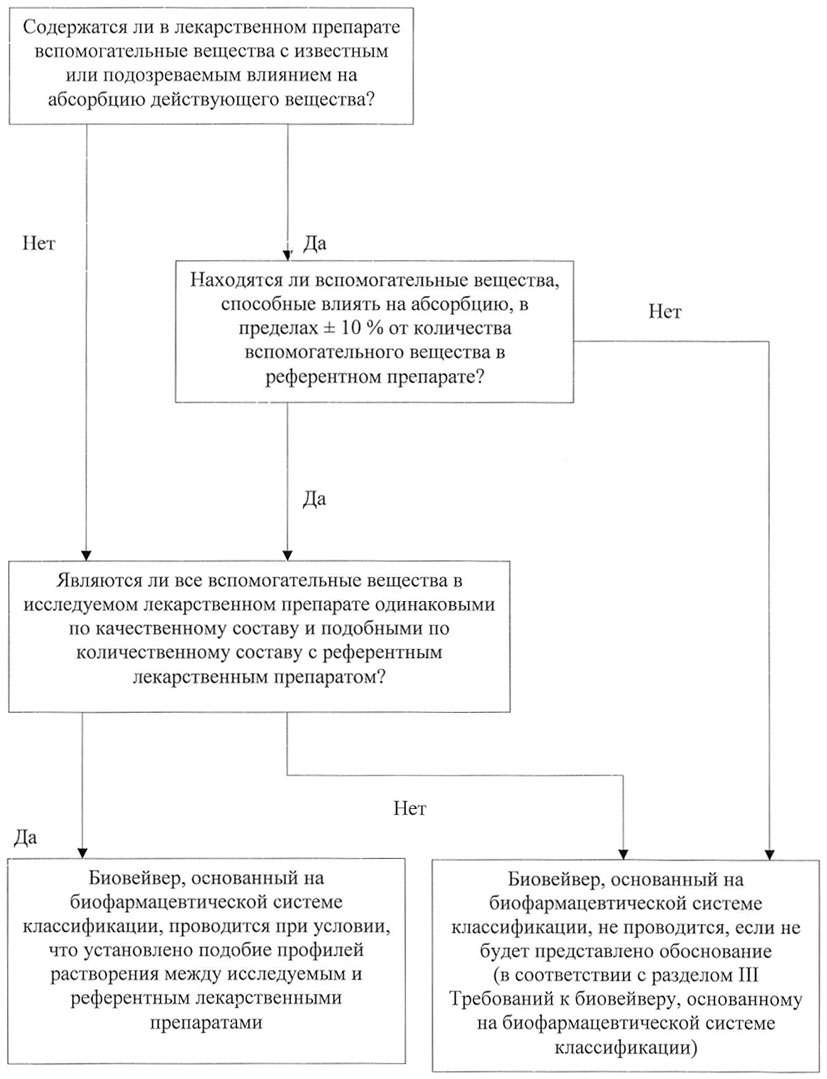
Рисунок 2. Алгоритм оценки лекарственного препарата,
содержащего действующие вещества класса III
по биофармацевтической системе классификации
2. Примеры допустимых различий в составе вспомогательных
веществ в исследуемом и референтном лекарственных препаратах
Пример 1. Биовейвер, основанный
на биофармацевтической системе классификации,
для лекарственного препарата, содержащего действующее
вещество класса I по биофармацевтической
системе классификации
Качественный состав исследуемого лекарственного препарата совпадает с качественным составом референтного лекарственного препарата. Количественное содержание сорбитола (вспомогательного вещества с известным или подозреваемым влиянием на абсорбцию действующего вещества) отличается от его содержания в референтном лекарственном препарате. При этом количественное содержание сорбитола в исследуемом препарате находится в допустимом диапазоне (45 - 55 мг), рассчитанном на основании содержания сорбитола в референтном препарате (50 мг ![]() 10%).
10%).
Компонент состава
Функциональное назначение
Содержание в референтном препарате (мг)
Содержание в исследуемом лекарственном препарате (мг)
Действующее вещество
100
100
Микрокристаллическая целлюлоза
наполнитель
100
95
Сорбитол
наполнитель
50
55
Гидроксипропилметил-целлюлоза
связующее вещество
10
10
Тальк
скользящее вещество
5
5
Итого
265
265
Пример 2. Биовейвер основанный
на биофармацевтической системе классификации,
для лекарственного препарата, содержащего действующее
вещество класса III по биофармацевтической
системе классификации
Качественный состав исследуемого лекарственного препарата совпадает с качественным составом референтного лекарственного препарата. Количественное содержание сорбитола (вспомогательного вещества с известным или подозреваемым влиянием на абсорбцию действующего вещества) отличается от его содержания в референтном лекарственном препарате. При этом количественное содержание сорбитола в исследуемом препарате находится в допустимом диапазоне 9 - 11 мг, рассчитанном на основании количественного содержания сорбитола в референтном лекарственном препарате (10 мг ![]() 10%). Различия в количественном содержании других вспомогательных веществ находятся в пределах границ, указанных в приведенной в пункте 35 Требований к биовейверу, основанному на биофармацевтической системе классификации таблице.
10%). Различия в количественном содержании других вспомогательных веществ находятся в пределах границ, указанных в приведенной в пункте 35 Требований к биовейверу, основанному на биофармацевтической системе классификации таблице.
Компонент состава
Функциональное назначение
Референтный препарат
Исследуемый препарат
Абсолютная разница относительно массы ядра таблетки
состав (мг)
доля от массы ядра таблетки (% м/м)
состав (мг)
доля от массы ядра таблетки (% м/м)
Действующее вещество
-
100,0
49,3%
100,0
46,5%
-
Лактозы моногидрат
наполнитель
85,0
41,9%
97,0
45,1%
3,2%
Сорбитол
наполнитель
10,0
4,9%
9,0
4,2%
0,7%
Кроскармеллоза натрия
дезинтегрант
6,0
3,0%
7,0
3,3%
0,3%
Магния стеарат
лубрикатор
2,0
1,0%
2,0
0,9%
0,1%
Итого
203,0
100,0%
215,0
100,0%
Общее отличие:
4,3%
Приложение N 3
к Требованиям к биовейверу,
основанному на биофармацевтической
системе классификации
ДОПОЛНЕНИЯ
К УСЛОВИЯМ ПРИМЕНЕНИЯ ТРЕБОВАНИЙ К БИОВЕЙВЕРУ, ОСНОВАННОМУ
НА БИОФАРМАЦЕВТИЧЕСКОЙ СИСТЕМЕ КЛАССИФИКАЦИИ
Положение Требований
Условия применения
Общие положения
применимость биовейвера, основанного на биофармацевтической системе классификации, к действующим веществам, имеющим нелинейную фармакокинетику
к действующим веществам, имеющим нелинейную фармакокинетику, допускается применять биовейвер, основанный на биофармацевтической системе классификации, если они отвечают критериям растворимости и проникающей способности для класса I или класса III по биофармацевтической системе классификации (далее - БКС).
допустимость применения биовейвера, основанного на БКС, для комбинированных лекарственных препаратов, в составе которых только одно из действующих веществ соответствует критериям биовейвера, основанного на биофармацевтической системе классификации, а остальные вещества - не соответствуют
биовейвер, основанный на БКС, допускается, если все действующие вещества комбинированного лекарственного препарата отвечают критериям класса I или класса III по БКС. Если хотя бы одно из действующих не является действующим веществом класса I или класса III по БКС, нельзя исключить возможность того, что лекарственная форма и состав комбинированного лекарственного препарата повлияют на поведение этого действующего вещества in vivo.
причина исключения лекарственных препаратов с узким терапевтическим индексом из сферы применения биовейвера, основанного на БКС, несмотря на зависимость скорости и степени абсорбции таких действующих веществ класса I или класса III по БКС только от их растворимости и проникающей способности и отсутствие зависимости от узкого терапевтического индекса
лекарственные препараты с узким терапевтическим индексом - это препараты, для которых небольшие различия в дозе или концентрации в крови могут привести к появлению зависящих от дозы или концентрации серьезных проблем (терапевтической неэффективности или нежелательных реакций). Для этих лекарственных препаратов характерен крутой вид зависимости "доза - ответ" в обычном дозовом диапазоне или узкий диапазон между эффективными концентрациями действующего вещества и концентрациями, связанными с серьезной токсичностью. Таким образом, дозы этих лекарственных средств необходимо осторожно титровать и мониторировать. Несмотря на отсутствие лекарственных препаратов с узким терапевтическим индексом, к подтверждению in vivo биоэквивалентности указанных лекарственных препаратов предъявляются особые требования, (например, более строгие критерии приемлемости (90% доверительный интервал для Cmax и (или) AUC должен находиться в диапазоне 90,00 - 111,00%) и определенные особенности дизайна исследования). Принципы биовейвера, основанного на БКС, не позволяют устанавливать более строгие критерии биовейвера. В связи с этим биовейвер, основанный на БКС, не является пригодным исследованием in vitro для установления биоэквивалентности лекарственных препаратов с узким терапевтическим индексом.
Критерии применения биовейвера, основанного на БКС
допустимость применения биовейвера, основанного на БКС, в случае, если исследуемый и референтный препараты содержат разные формы солей одного и того же действующего вещества
биовейвер, основанный на БКС, допускается применять, если исследуемый и референтный лекарственные препараты содержат разные (простые) соли действующего вещества при условии, что обе соли принадлежат к классу I по БКС (обладают хорошей растворимостью и хорошей проникающей способностью).
Биовейвер, основанный на БКС, не применяется, если исследуемый лекарственный препарат содержит действующее вещество, которое по своему строению (простой эфир, сложный эфир, изомер, смесь изомеров, комплексное соединение или иное химическое производное) отличается от строения действующего вещества референтного препарата, поскольку такие различия могут привести к различиям в биодоступности, которые не возможно установить с помощью экспериментов, используемых для обоснования биовейвера, основанного на БКС. Помимо научных аспектов обоснования возможности применения биовейвера, основанного на БКС, необходимо учитывать требования Правил проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза.
необходимость учета изменения массы активной фармацевтической субстанции, связанного с иной солью действующего вещества при оценке растворимости
в БКС классифицируется непосредственно действующее вещество (основание). Доза конкретного действующего вещества в эксперименте должна быть идентичной независимо от вида его соли. Следовательно, понятие "отличия массы действующего вещества" в данной ситуации неприменимо.
обоснование применения биовейвера, основанного на БКС, к пролекарствам, которые всасываются только в виде пролекарства
БКС основана на критериях растворимости и проникающей способности конкретного действующего вещества. Классификационная группа не может быть перенесена на другие соединения, например, на исходное вещество или метаболит. Оценка критерия растворимости проводится исходя из предположения, что лекарственный препарат принимается внутрь с определенным количеством воды. Это условие не применимо для метаболита, если только он не образуется непосредственно сразу после приема и до абсорбции. БКС применяется к действующему веществу в составе лекарственного препарата, поскольку для подтверждения схожести препаратов используется in vitro растворение этого действующего вещества.
Растворимость
поддержание постоянства pH во время эксперимента по оценке растворимости
допускаются различные методы поддержания pH раствора. Если требуется поддержание постоянства pH, необходимо обосновать выбранный метод. Отклонение pH в пределах ![]() 0,1 считается приемлемым.
0,1 считается приемлемым.
определение продолжительности установления растворимости
в случае оценки равновесной растворимости продолжительность установления растворимости должна быть научно обоснована исходя из времени, необходимого для достижения равновесного состояния. В случаях, когда равновесную растворимость определить невозможно, продолжительность эксперимента растворимости должна быть научно обоснована с учетом ожидаемого времени абсорбции in vivo.
учет влияния ионной силы буферных растворов при испытании на растворимость
ионная сила буферных растворов не влияет на растворимость действующих веществ.
допустимость использования средних значений повторов оценки растворимости или наименьшего из полученных значений растворимости при значительной вариабельности показателей между отдельными результатами определения растворимости при заданном pH
в отношении хорошо растворимых действующих веществ не должна возникать существенная вариабельность между значениями индивидуальных определений растворимости. Однако при наличии вариабельности определение растворимости должно основываться на среднем значении повторов оценки растворимости.
допустимость использования данных научной литературы или иных научных данных для обоснования растворимости в качестве опорных данных для отнесения действующего вещества в соответствующий класс БКС
для установления растворимости действующего вещества необходимо представить экспериментальные данные о растворимости. Научные химикотехнологические и фармацевтические данные представляются только для дальнейшего подтверждения выводов о растворимости.
допустимость использования при оценке растворимости иного предела для деградации действующего вещества, по сравнению с установленным Требованиями к биовейверу, основанному на биофармацевтической системе классификации (далее - Требования) пределом не более 10%
предел деградации действующего вещества не более 10% установлен для обеспечения того, чтобы не происходило завышение показателя растворимости, связанного с переходом вещества в раствор, но не с деградацией этого действующего вещества. Указанный предел может быть обеспечен в условиях эксперимента.
Проникающая способность
допустимость использования иных полностью валидированных клеточных линий (например, MDCKII, LLC-PK1) вместо клеточной линии Caco-2, для получения оценки проникающей способности действующего вещества по БКС
проникающую способность действующего вещества принципиально можно оценить с помощью других методов in vitro (иные клеточные линии, например, MDCKII), in situ (Loc-1-Gut) или ex vivo (модель вывернутого мешка, сформированного из кишечника крысы). Поскольку опыт оценки проникающей способности действующих веществ с помощью in vitro подходов незначительный, необходимо применять метод, для которого накоплен наибольший опыт применения, которым является клеточная линия Caco-2. По мере накопления экспериментальных данных в отношении иных методов ex vivo и in situ на основе клеточных линий или животных, эти данные допускается использовать в последующем при наличии строгой валидации и стандартизации в соответствии с принципами, изложенными в приложении N 1 к Требованиям.
причины классификации действующих веществ, проявляющих умеренную проникающую способность (50 - 84%) в валидированных исследованиях на клеточной линии Caco-2 и являющихся нестабильными в желудочно-кишечном тракте в группу действующих веществ имеющих плохую проникающую способность
только действующим веществам с хорошей проникающей способностью присваивается класс I по БКС (что обеспечивает дополнительную гибкость для производителя при изменении состава вспомогательных веществ и дает возможность применения более широких критериев для растворения (![]() 85% в течение 30 минут)).
85% в течение 30 минут)).
Отнесение к данному классу действующих веществ с проникающей способностью, отличной от хорошей проникающей способности (то есть веществ с умеренной или плохой проникающей способностью), не допускается в контексте биовейвера, основанного на биофармацевтической системе классификации. В отношении действующих веществ, не стабильных в желудочно-кишечном тракте, невозможно подтвердить хорошую проникающую способность in vivo. В случаях, когда хорошую проникающую способность невозможно подтвердить однозначно с помощью одного из методов, описанных в Требованиях, применение биовейвера, основанного на БКС, возможно если действующее вещество отвечает критериям класса III по БКС (лекарственный препарат имеет ограничения в отношении изменений состава и содержания вспомогательных веществ и является очень быстро растворимым (происходит высвобождение ![]() 85% действующего вещества в течение 15 минут)).
85% действующего вещества в течение 15 минут)).
обеспечение размера выборки, необходимого для получения надежной оценки проникающей способности действующего вещества
планируемое количество повторов исследований, необходимое для правильного определения класса проникающей способности действующего вещества, трудно однозначно определить, поскольку оно зависит от вариабельности каждого отдельного анализа. Для этой группы анализов характерна высокая межлабораторная вариабельность, для которой в научной медицинской и химикофармацевтической литературе (J Pharm Sci (97), 2008; Eur J Pharm&Biopharm (114), 2017)) установлены потенциальные источники вариабельности. Межлабораторная вариабельность существенно ниже в случае принадлежности действующего вещества к классу I по сравнению с классом III по БКС. В отношении действующих веществ с 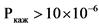 см/с имеются данные об умеренной вариабельности (5 - 20%), представленные в научной медицинской и химико-фармацевтической литературе Eur J Pharm Sci (56), 2014; J Pharmcol & Toxicol Methods (70), 2014. В связи с этим маловероятно, что высокая вариабельность приведет к неправильной оценке хорошей проникающей способности действующего вещества. Для анализов с использованием монослоев эпителиальных клеток линии Caco-2 минимально обоснованным является выполнение 3 повторов исследований.
см/с имеются данные об умеренной вариабельности (5 - 20%), представленные в научной медицинской и химико-фармацевтической литературе Eur J Pharm Sci (56), 2014; J Pharmcol & Toxicol Methods (70), 2014. В связи с этим маловероятно, что высокая вариабельность приведет к неправильной оценке хорошей проникающей способности действующего вещества. Для анализов с использованием монослоев эпителиальных клеток линии Caco-2 минимально обоснованным является выполнение 3 повторов исследований.
подходы к статистической дифференцировке полученных значений Ркаж для лекарственных препаратов с плохой, умеренной и хорошей проникающей способностью в случае, если значения Ркаж накладываются друг на друга при сравнении отдельных значений для лекарственных препаратов из каждой группы
целью статистической дифференцировки экспериментальных значений является получение дихотомичного результата (действующее вещество проявляет или не проявляет хорошую проникающую способность). In vzvo-проникающая способность референтных действующих веществ, перечисленных в приложении N 1 к Требованиям, подтверждена в исследованиях с участием человека, которые показывают, что средние значения проникающей способности этих действующих веществ четко позволяют их разделить на вещества с плохой, умеренной и хорошей проникающей способностью. Экспериментальные системы на основе линии клеток Caco-2 позволяют провести их валидацию для целей определения класса исследуемого действующего вещества по БКС, используя указанные референтные действующие вещества. Для успешной валидации необходимо получить дифференцированные результаты с использованием действующих веществ с хорошей, умеренной и плохой проникающей способностью in vitro.
Если средние значения для действующих веществ с плохой, умеренной и хорошей проникающей способностью накладываются друг на друга при экспериментальном определении, то это указывает на вероятные проблемы постановки или выполнения методики исследования с использованием клеточной линии Caco-2 в организации. Для подтверждения класса проникающей способности исследуемого действующего вещества анализ стандартизуется по указанным референтным действующим веществам. Исследуемое действующее вещество классифицируют как вещество с хорошей проникающей способностью если его кажущаяся проникающая способность (Ркаж) равна или превышает такую способность у референтного действующего вещества с хорошей проникающей способностью. В этом случае дальнейшей статистической дифференцировки результатов проводить не требуется.
Пригодность лекарственного препарата для биовейвера, основанного на БКС
ограничения в использовании разных лекарственных форм исследуемого и референтного лекарственного препаратов для биовейвера, основанного на БКС
различия в лекарственных формах одного и того же действующего вещества могут влиять на его поведение in vivo. Конкретные указания применительно к разным лекарственным формам и вспомогательным веществам рассмотрены в Требованиях, в том числе с учетом влияния различий, привнесенных лекарственной формой, на поведение действующего вещества in vivo и в целях уменьшения риска, связанного с вероятностью сделать неправильное заключение (вывод) о биоэквивалентности. Положения Требований допускается применять для замены одной лекарственной формы на другую во время разработки лекарственного препарата, если это обосновано, например, с помощью предыдущих данных in vivo.
ограничения в применении биовейвера, основанного на БКС, для диспергируемых во рту таблеток, при условии, что они применяются без запивания водой
поскольку остаточный объем желудка значительно меньше 250 мл, оценка растворимости действующего вещества в 250 мл среды растворения не применима к лекарственным препаратам, принимаемым без запивания водой. Определение объема среды растворения, требуемого для установления класса растворимости, будет затруднительным для диспергируемых во рту таблеток, при условии, что они применяются без запивания водой. Текущая методика изучения растворения ограниченно пригодна для лекарственного препарата, диспергирование которого происходит в ротовой полости без приема стакана воды. В отношении таких лекарственных препаратов необходимо проводить исследование биоэквивалентности с диспергируемыми во рту таблетками, при условии, что они не запиваются водой.
Вспомогательные вещества
допустимость использования методики in silico физиологически обоснованного фармакокинетического моделирования абсорбции для оценки риска изменения поведения лекарственной формы, связанной с потенциальным влиянием (включением или исключением) изменения вспомогательного вещества сверх рекомендуемых диапазонов
физиологически обоснованное фармакокинетическое моделирование абсорбции in silico используется для оценки риска в поведении препарата вследствие изменения лекарственной формы. Однако в настоящее время подобные модели не могут исчерпывающе предсказывать все потенциальные различия в абсорбции, опосредуемые критичными вспомогательными веществами. Валидация моделей in silico для подобных целей ограничена в связи с отсутствием понимания механизмов некоторых наблюдаемых эффектов вспомогательных веществ и отсутствием качественных данных in vivo для некоторых классов вспомогательных веществ. В связи с этим риск оценки неверной оценки на основании предсказанных моделью эффектов не позволяет обосновывать изменение вспомогательного вещества сверх рекомендуемого диапазона. В некоторых случаях in silico физиологически обоснованное фармакокинетическое моделирование позволяет получить необходимое обоснование в рамках более широкой оценки рисков вспомогательных веществ, например, при анализе чувствительности с использованием должным образом валидированного физиологически обоснованного фармакокинетического моделирования абсорбции для вспомогательных веществ, когда механизм эффекта хорошо понятен.
влияние вспомогательных веществ, указанных в пункте 35 Требований как "прочие вспомогательные вещества" на абсорбцию
приведенная в пункте 35 Требований таблица, содержит критерии для подтверждения количественного подобия лекарственных препаратов, содержащих действующие вещества класса III по БКС. Перечисленные в таблице классы вспомогательных веществ являются функциональными классами, однако в пределах каждого класса вспомогательное вещество может повлиять на абсорбцию. В этом случае процентная разница в количестве такого вспомогательного вещества по сравнению с его содержанием в референтном лекарственном препарате должна укладываться в 10% отклонения от массы этого вещества.
данные, которые рассматриваются как "соответствующее обоснование" для отклонения от приемлемой разницы во вспомогательных веществах, указанных в приведенной пункте 35 Требований таблице
такими данным являются результаты относительно in vivo поведения лекарственной формы, полученные во время программы разработки лекарственного препарата. Такие данные (например, для лекарственных форм с разными диапазонами содержания вспомогательных веществ, не проявляющих влияния на абсорбцию действующего вещества (включая подробную оценку механизма их эффектов)), допускается использовать в качестве обоснования изменения содержания вспомогательных веществ, сверх пределов, указанных в приведенной в пункте 35 Требований таблице.
допустимость использования в составе лекарственного препарата вспомогательных веществ одинаковой разновидности с веществами в составе референтного лекарственного препарата, но другого функционального класса, как обеспечения требования "качественно подобного состава" в отношении действующих веществ класса III по БКС
если это обосновано, разницу в функциональном классе вспомогательного вещества необходимо оценивать относительно его функциональных свойств непосредственно в составе промышленной рецептуры и лекарственной формы конкретного лекарственного препарата. В отношении некоторых разновидностей вспомогательных веществ не имеется доказательств влияния изменения функционального класса вспомогательного вещества на поведение лекарственного препарата в организме человека. В отношении других вспомогательных веществ модификация класса может потенциально повлиять на растворение лекарственного препарата (например, изменение в распределении по размеру частиц, изменение вязкости, и (или) степени замещения гидроксипропилметилцеллюлозы, изменение удельной площади поверхности лубрикаторов из группы стеаратов). Оценка сопоставимости вспомогательных веществ требует принятия решений о допустимости такой сопоставимости в индивидуальном порядке, чтобы однозначно подтвердить сохранение качественного подобия состава.
приемлемые границы отличий в содержании вспомогательных веществ из группы многоатомных спиртов
в настоящее время отсутствуют достаточные данные для квалификации порогов, при которых влияние этих вспомогательных веществ на поведение лекарственного препарата становится значимым. Более того, влияние изменений, вызываемое такими вспомогательными веществами, будет зависеть от свойств действующего вещества (чувствительности фармакокинетического профиля действующего вещества к изменению кишечного транзита). Изменения в содержании вспомогательных веществ этой группы должны соответствовать тем же ограничениям, которые применяются к другим вспомогательным веществам, способным влиять на абсорбцию действующего вещества, то есть находиться в пределах ![]() 10% от количества вспомогательного вещества в референтном лекарственном препарате.
10% от количества вспомогательного вещества в референтном лекарственном препарате.
примеры составов лекарственных препаратов, содержащих действующие вещества класса III по БКС, для которых у вспомогательных веществ выполняется и не выполняется критерий совпадения качественного состава и сходства количественного состава
примеры таких лекарственных препаратов, сохраняющих сходство количественных составов вспомогательных веществ, приведены в приложении N 2 к Требованиям. Дополнительные указания по допустимым различиям состава вспомогательных веществ представлены в таблице примера 2 приложения N 2 к Требованиям в виде разницы в процентах относительно массы ядра таблетки (массовой доли). Если исследуемый лекарственный препарат соответствует этим указаниям, но имеет большие различия в абсолютных количествах вспомогательных веществ (например, если масса ядра исследуемого и референтного лекарственного препарата не схожа), уполномоченный орган (экспертная организация) государства - члена Евразийского экономического союза вправе истребовать дополнительное обоснование.
Тест сравнительной кинетики растворения in vitro для лекарственного препарата
допустимость использования грузил (синкеров) в тесте сравнительной кинетики растворения не только при появлении феномена конусообразования, но и в иных случаях (например, при прилипании частиц лекарственной формы к лопастной мешалке, их флотации и т.д.)
использование грузил (синкеров) для устранения проблем, зафиксированных во время экспериментов по растворению, допускается при наличии обоснования. При этом к исследуемой и референтной лекарственным формам необходимо применять одни и те же экспериментальные условия.
подход к сравнению профилей растворения для лекарственных препаратов класса I по БКС, если один профиль растворения соответствует критериям для очень быстрого растворения (![]() 85% для среднего количества вещества, перешедшего в раствор за
85% для среднего количества вещества, перешедшего в раствор за ![]() 15 минут), а другой профиль растворения соответствует критериям для быстрого растворения (
15 минут), а другой профиль растворения соответствует критериям для быстрого растворения (![]() 85% для среднего количества вещества, перешедшего в раствор за < 30 минут) характеристикам профиля растворения in vitro
85% для среднего количества вещества, перешедшего в раствор за < 30 минут) характеристикам профиля растворения in vitro
если один препарат показывает растворение более чем 85% за 15 минут, а второй - нет, то необходимо предусмотреть достаточное количество точек взятия образцов, чтобы рассчитать показатель f2 для подтверждения подобия профилей растворения.
необходимость расчета по временным точкам отбора проб параметра f2 для сравнения профилей растворения в случае если они различаются между исследуемым и референтным лекарственными препаратами (быстрое и очень быстрое растворение)
в этих случаях по временным точкам, в которых был отбор проб должен быть рассчитан параметр f2. Если один препарат показывает растворение более чем 85% за 15 минут, а второй - нет, необходимо предусмотреть достаточное количество точек взятия образцов, чтобы рассчитать показатель f2 для подтверждения подобия профилей растворения.
допустимость использования при сравнений профилей растворения недостаточного количества точек взятия образцов для расчета показателя f2, если это недостаточное количество связано с исключением ранних временных точек вследствие высокой вариабельности
в отношении действующих веществ I класса по БКС высокая вариабельность по растворению не ожидается, поэтому альтернативные статистические методологии (например, бутстрэппинг (бутстреп анализ)), для подтверждения подобия профилей растворения не считаются применимыми. В случае если высокая вариабельность возникает вследствие феномена конусообразования, для решения таких проблем рассматриваются альтернативные методики выполнения исследования растворения (например, использование грузил (синкеров) или других обоснованных подходов), если это обосновано с научной точки зрения.
подход к сравнению профилей растворения в случае, если выбор разных временных точек отбора проб приводит к разным значениям параметра f2, требующим взаимопротиворечащих выводов (например, временные точки 10, 20, 30 минут приводят к значению f2 < 50, а временные точки 8, 20, 30 минут приводят к значению f2 > 50)
подобная ситуация должна возникать только в исключительных случаях. Временные точки для расчета параметра f2 должны быть предустановлены. При расчете параметра необходимо использовать все отвечающие критериям приемлемости предустановленные временные точки взятия образцов и выбор этих временных точек должен быть обоснован.
допустимость распространения биовейвера, основанного на БКС, для одной дозировки препарата на другие дозировки из линейки дозировок лекарственного препарата
подобное заключение недопустимо. Биовейвер, основанный на БКС, требует представления обосновывающих данных для каждой отдельной дозировки из линейки дозировок лекарственного препаратов. Сравнение in vitro дозировок исследуемого лекарственного препарата с соответствующими дозировками референтного лекарственного препарата исключает возможный дрейф результатов подобия, который может возникнуть, если оценка высвобождения дополнительного количества действующего вещества в дозировке выполняется без сравнения с соответствующей дозировкой референтного лекарственного препарата.
допустимость сравнения между следующими лекарственными формами для заявления на биовейвер, основанный на БКС:
а) не покрытые оболочкой таблетки в сравнении с таблетками, покрытыми пленочной оболочкой;
б) таблетки в сравнении с капсулами
а) не покрытые оболочкой таблетки и таблетки, покрытые пленочной оболочкой, не выполняющей определенное функциональное предназначение, рассматриваются в качестве одной и той же лекарственной формы при сравнении по биовейверу. Сравнение между указанными формами дозирования допускается для биовейвера, основанного на БКС;
б) таблетки и капсулы не рассматриваются в качестве одной и той же лекарственной формы сравнения по биовейверу. Сравнение между указанными формами дозирования не допускается для биовейвера, основанного на БКС.
требуемые показатели скорости перемешивания среды для сравнительной оценки кинетики растворения для суспензионных лекарственных форм
в отношении суспензий рекомендуемая скорость вращения лопастей равна 50 об/мин в Приборе 2 (лопастная мешалка). Допускается (но необязательно) использовать меньшую скорость вращения лопастей.
Методика анализа проникающей способности действующего вещества с использованием линии клеток Caco-2
допустимость использования для валидации метода на клетках Caco-2 при оценке класса действующего вещества по БКС за счет установления in vitro проникающей способности путем пассивной диффузии указанных в таблице приведенной в приложении N 1 к Требованиям, 12 модельных действующих веществ, которые подвергаются активному транспорту (4 из 12 модельных действующих веществ подвергаются активному эффлюксу (дигоксин, паклиткасел, хинидин и винбластин), остальные 8 веществ подвергаются активному транспорту (фуросемид - переносчиком OAT3, метформин - переносчиками OCT1 и OCT2, амилорид - переносчиком OCT2, фамотидин - переносчиком OCT2, ацикловир - переносчиками OAT1 и OCT1, теофиллин - переносчиком OAT2 и эналаприл переносчиками PepT1 и PepT2)
результаты сравнения проникающих способностей 24 действующих веществ, представленные в научной медицинской и химико-фармацевтической литературе (Pharm Res (19) 2002 и Drug Discover Today (17) 2012) и полученные на тощей кишке человека и в линии клеток Caco-2 in vivo и in vitro, имели хорошую корреляцию для веществ абсорбируемых пассивной диффузией и несколько худшую для активного транспорта. Поэтому монослои клеток Caco-2 могут использоваться для прогнозирования пассивной диффузии действующих веществ у человека, тогда как прогнозирование активного транспорта с помощью систем переносчиков может быть менее точным, учитывая измененную экспрессию переносчиков этой клеточной линией. Соответственно модельные лекарственные препараты, для определения хорошей проникающей способности, являются быстро (пассивно) проникающими лекарственными препаратами, такими как напроксен, антипирин и метопролол с сопоставимыми коэффициентами проникающей способности на клетках Caco-2 и в тощей кишке человека. Несмотря на то, что некоторые модельные лекарственные препараты частично подвергаются активному транспорту, проникающие способности их действующих веществ на монослоях клеток линии Caco-2 имеют надежную корреляцию с проникающей способностью in vivo. Поскольку экспрессия переносчиков клеточными линиями может отличаться от условий in vivo, эта корреляция не является универсальной для всех действующих веществ переносимых активным транспортом. В связи с этим без представления доказательных in vivo данных in vitro данные не являются единственным способом определения класса проникающей способности действующих веществ, переносимых активным транспортом. Окончательный вывод о действующем веществе, классифицируемом как хорошо проникающее действующее вещество с помощью анализа на монослое клеток линии Caco-2, будет возможен только для действующих веществ, не переносимых никаким активным транспортом.
допустимость использования биовейвера, основанного на БКС, для действующих веществ с низкими показателями Km, если выполняются все следующие условия:
а) данные in vitro для действующего вещества подвергаемого эффлюксу в клетках линии Caco-2, позволяют получить кажущееся значение Km значительно ниже, чем соответствующие концентрации этого действующего вещества в кишечнике;
б) активность процесса эффлюкса достигает насыщения при всех концентрациях действующего вещества, а проникающая способность действующего вещества определяется только его пассивной диффузией;
в) клиническая фармакокинетика действующего вещества человека является линейной;
г) представлены вспомогательные данные, например данные по фармакокинетике (абсорбции, распределении, метаболизме, элиминации) у человека
отсутствие эффлюкса или насыщение эффлюкс-переносчиков невозможно экспериментально различить, если используемые физиологически обоснованные концентрации (например, в соответствии с приложением N 1 к Требованиям в 0,01, 0,10 и 1,00 от наибольшей дозировки лекарственного препарата, растворенной в 250 мл среды) превышают значение Km действующего вещества. В этом случае действующее вещество считается хорошо проникающим, если кажущаяся проникающая способность (Ркаж) больше или равна Ркаж референтного модельного лекарственного препарата с большой проникающей способностью. Анализ с использованием клеток Caco-2 при этом должен быть валидирован, подтверждая двунаправленный характер транспорта известных модельных лекарственных препаратов (таблица, приведенная в приложении N 2 к Требованиям) и доказывать функциональную активность эффлюкс-переносчика (переносчиков). Если при этом возможно представить in vivo данные, показывающие хорошую проникающую способность действующего вещества в соответствии с настоящими Требованиями (то есть данные по фармакокинетике (абсорбции, распределении, метаболизме, элиминации) у человека или абсолютную биодоступность), класс хорошей проникающей способности для действующего вещества также может быть присвоен. Необходимо учитывать, что для действующих веществ класса III по БКС, не соответствующих критериям класса хорошей проникающей способности, вариант биовейвера, основанного на БКС также доступен, если выполняются все условия в соответствии с Требованиями.
допустимость исключения из рассмотрения по биовейверу, основанному на БКС, транспортируемых действующих веществ для которых клетки линии Caco-2 не позволяют надежно классифицировать проникающую способность
к активно транспортируемым действующим веществам применимы условия, описанные для действующих веществ с низкими показателями Km. Активно транспортируемые действующие вещества не исключаются из рассмотрения по биовейверу, основанному на БКС, если in vivo данные полученные в исследованиях с участием человека, позволяют обосновать принадлежность действующего вещества к классу веществ с хорошей проникающей способностью. Для данного случая не допускается исключительное использование анализа проникающей способности методом ее изучения на клетках Caco-2 (поскольку экспрессия переносчиков системами Caco-2 может отличаться от экспрессии in vivo).
допустимость обоснования порога эффлюкс-отношения больше 2 с помощью модельных соединений (наборов данных из валидационных результатов) в случае отдельных валидированных моделей монослоев клеток Caco-2 для которых эффлюкс-отношение больше 2 является более приемлемым
в отсутствие какого-либо активного транспорта (абсорбции или эффлюкса), отношение между абсорбционным Ркаж (в направлении от апикального к базолатеральному или "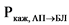 ") и Ркаж "
") и Ркаж "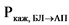 " должно быть равно 1 или быть близким к такому значению. Любое отклонение от 1 будет указывать на некоторый вклад активного транспорта в процесс переноса действующего вещества. Эффлюкс-отношение больше 2 принято в качестве порога, указывающего на то, что действующее вещество является субстратом эффлюкс-переносчика.
" должно быть равно 1 или быть близким к такому значению. Любое отклонение от 1 будет указывать на некоторый вклад активного транспорта в процесс переноса действующего вещества. Эффлюкс-отношение больше 2 принято в качестве порога, указывающего на то, что действующее вещество является субстратом эффлюкс-переносчика.
работы, в которых приводятся данные по выбору модельных лекарственных препаратов для валидации метода оценки проникающей способности действующих веществ
соответствующие данные приведены в следующих работах:
Volpe DA. Application of Method Suitability for Drug Permeability Classification. AAPS J. 2010; 12(4):670 - 8;
Li C. et al. Development of In Vitro Pharmacokinetic Screens Using Caco-2, Human Hepatocyte, and Caco-2/Human Hepatocyte Hybrid Systems for the Prediction of Oral Bioavailability in Humans. Journal of Biomolecular Screening 2007; 12(8):1084 - 1091;
Peng Y. et al. Applications of a 7-day Caco-2 cell model in drug discovery and development. European Journal of Pharmaceutical Sciences 2014; 56: 120 - 130;
Kasim NA et al. Molecular Properties of WHO Essential Drugs and Provisional Biopharmaceutical Classification. Molecular Pharmaceutics 2004; 1(1): 85 - 96;
Lennernas, H. Intestinal permeability and its relevance for absorption and elimination, Xenobiotica 2007; 37(10): 1015 - 1051;
Thiel-Demby VE. Biopharmaceutics Classification System: Validation and Learnings of an In Vitro Permeability Assay. Molecular Pharmaceutics 2009; 6(1): 11 - 18;
Giacomini, et al. Nat Rev Drug Discov. 2010; 9:215 - 236;
FDA, United States In Vitro Metabolism-and Transporter-Mediated Drug-Drug Interaction Studies Guidance for Industry (October 2017).
".
5. Пункты 9 и 10 приложения N 5 изложить в следующей редакции:
"9. Сопоставимость профилей растворения определяется в соответствии с пунктом 41 приложения N 4 к Правилам проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза, утвержденным Решением Совета Евразийской экономической комиссией от 3 ноября 2016 г. N 85.
10. Расчет фактора подобия основан на выполнении всех условий в соответствии с пунктом 42 приложения N 4 к Правилам проведения исследований биоэквивалентности лекарственных препаратов в рамках Евразийского экономического союза, утвержденным Решением Совета Евразийской экономической комиссией от 3 ноября 2016 г. N 85.".