Решение Коллегии Евразийской экономической комиссии от 15.09.2020 N 111
КОЛЛЕГИЯ ЕВРАЗИЙСКОЙ ЭКОНОМИЧЕСКОЙ КОМИССИИ
РЕШЕНИЕ
от 15 сентября 2020 г. N 111
ОБ УТВЕРЖДЕНИИ РУКОВОДСТВА
ПО ФАРМАКОКИНЕТИЧЕСКОМУ И КЛИНИЧЕСКОМУ ИЗУЧЕНИЮ
БИОЭКВИВАЛЕНТНОСТИ ЛИПОСОМАЛЬНЫХ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ
ДЛЯ ВНУТРИВЕННОГО ВВЕДЕНИЯ
В соответствии со статьей 30 Договора о Евразийском экономическом союзе от 29 мая 2014 года, статьей 6 Соглашения о единых принципах и правилах обращения лекарственных средств в рамках Евразийского экономического союза от 23 декабря 2014 года Коллегия Евразийской экономической комиссии решила:
1. Утвердить прилагаемое Руководство по фармакокинетическому и клиническому изучению биоэквивалентности липосомальных лекарственных препаратов для внутривенного введения.
2. Настоящее Решение вступает в силу по истечении 6 месяцев с даты его официального опубликования.
Председатель Коллегии
Евразийской экономической комиссии
М.МЯСНИКОВИЧ
Утверждено
Решением Коллегии
Евразийской экономической комиссии
от 15 сентября 2020 г. N 111
РУКОВОДСТВО
ПО ФАРМАКОКИНЕТИЧЕСКОМУ И КЛИНИЧЕСКОМУ ИЗУЧЕНИЮ
БИОЭКВИВАЛЕНТНОСТИ ЛИПОСОМАЛЬНЫХ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ
ДЛЯ ВНУТРИВЕННОГО ВВЕДЕНИЯ
I. Общие положения
1. Одна из стратегий разработки систем доставки липосомальных лекарственных препаратов для внутривенного введения (далее - липосомальные лекарственные препараты) с целью усовершенствования специфичной для каждого заболевания направленности действия, контроля скорости высвобождения действующего вещества и (или) получения лекарственной формы, пригодной для клинического применения, заключается во включении (инкапсулировании) действующего вещества (веществ) в водную фазу липосомы или в ее встраивании в липидный компонент (связывании). В классическом определении липосомы представляют собой искусственно созданные везикулы, которые состоят из одного или нескольких концентрических липидных бислоев, содержащих в себе одну или несколько водных камер (компартментов), и к которым относятся моно- и мультиламеллярные липосомы, мультивезикулярные липосомы и липосомы, покрытые полимерной оболочкой.
2. В любом лекарственном препарате часть действующего вещества может находиться вне липосом - в свободной форме в основном растворе.
3. Поскольку липосомальные лекарственные препараты обладают рядом критических фармакокинетических свойств (включая их быстрое распознавание и элиминацию моноцитарно-фагоцитарной системой организма и преждевременное высвобождение действующего вещества из липосомы (нестабильность)), такие физико-химические свойства липосом, как размер частиц, текучесть (жидкостность) мембраны, поверхностный заряд и состав способствуют возникновению подобных критических фармакокинетических свойств in vivo. Свойства некоторых липосомальных лекарственных препаратов улучшались после добавления стеролов (например, холестерола), уменьшения размера и модификации поверхности с помощью ковалентного связывания с полимерами (например, с полиэтиленгликолем).
4. В противоположность лекарственным препаратам, в которых действующее вещество находится в форме истинного раствора, липосомальные лекарственные препараты обладают характеристиками распределения после внутривенного введения и зависящими от состава и специфики процесса производства. Поэтому аналогичные концентрации их действующих веществ в плазме могут не коррелировать с терапевтической активностью. Даже в случаях, когда состав липосом идентичен, изменения в их производстве, контроле соблюдения технологии производства липосом и контроле качества готового лекарственного препарата могут приводить к различной терапевтической активности липосомальных лекарственных препаратов. Для того чтобы гарантировать безопасность и эффективность нового липосомального лекарственного препарата, следует установить в полном объеме характеристики его стабильности и фармакокинетики (включая распределение в тканях). Это обусловлено тем, что различия между воспроизведенным и референтным липосомальными лекарственными препаратами в части, касающейся стадий процесса производства и состава липосомального лекарственного препарата, могут значительно влиять на его безопасность и эффективность вследствие изменений характера взаимодействия "липосома - клетка" и распределения липосом по их физическим характеристикам, которые невозможно обнаружить с помощью обычных исследований биоэквивалентности. При составлении программы доклинических и клинических исследований липосомальных лекарственных препаратов, разрабатываемых в качестве аналогов конкретного референтного липосомального лекарственного препарата, следует учитывать цели разработки последнего, а также данные, обосновывающие его применение.
5. Референтный липосомальный лекарственный препарат, используемый в исследованиях сопоставимости, должен быть зарегистрирован в качестве липосомального лекарственного препарата на территориях государств - членов Евразийского экономического союза (далее - государства-члены), его следует использовать в качестве контроля во всех предлагаемых фармацевтических испытаниях показателей качества, а также в опорных доклинических и клинических исследованиях сопоставимости.
6. В настоящем Руководстве описаны принципы оценки липосомальных лекарственных препаратов, разрабатываемых в качестве аналогов референтных липосомальных лекарственных препаратов. Настоящее Руководство не содержит требований к выбору конкретной стратегии при проведении химико-фармацевтических доклинических или клинических исследований.
Поскольку в настоящее время имеются ограниченные данные о фармацевтической разработке воспроизведенных липосомальных лекарственных препаратов, в настоящем Руководстве приведены общие указания по подтверждению и оценке сопоставимости таких воспроизведенных липосомальных лекарственных препаратов референтным липосомальным лекарственным препаратам. По вопросам подтверждения сопоставимости отдельных видов липосомальных лекарственных препаратов следует обращаться за научной консультацией в соответствии с пунктом 26 Правил регистрации и экспертизы лекарственных средств для медицинского применения, утвержденных Решением Совета Евразийской экономической комиссии от 3 ноября 2016 г. N 78.
7. Применение настоящего Руководства позволит разработчику (производителю) липосомального лекарственного препарата получить необходимые данные о качестве, данные доклинических и клинических исследований для обоснования регистрации липосомальных лекарственных препаратов, разрабатываемых в качестве аналогов референтного липосомального лекарственного препарата. Настоящее Руководство содержит указания по следующим вопросам:
представление фармацевтических данных в составе регистрационного досье лекарственного препарата, необходимых для подтверждения сопоставимости воспроизведенного липосомального лекарственного препарата к референтному липосомальному лекарственному препарату или для подтверждения сопоставимости измененного липосомального лекарственного препарата к первоначальному липосомальному лекарственному препарату в целях обоснования сопоставимой безопасности и эффективности таких препаратов;
необходимость проведения доклинических и клинических исследований и обстоятельства, позволяющие отказаться от проведения определенных исследований;
дизайн релевантных доклинических исследований in vivo и потенциальная роль моделей in vitro.
8. Принципы, изложенные в настоящем Руководстве, также могут применяться к новым видам лекарственных препаратов, подобным липосомальным, и везикулярных лекарственных препаратов, которые могут находиться в разработке (включая пути введения, отличные от внутривенного). Лекарственные препараты на основе фосфолипидов, в которых не происходит высвобождение действующего вещества из фосфолипидной системы, то есть в которых такая система служит исключительно растворителем действующего вещества, не относятся к липосомальным или к лекарственным препаратам, подобным липосомальным, и в настоящем Руководстве не рассматриваются.
II. Определения
9. Для целей настоящего Руководства используются понятия, которые означают следующее:
"несвязанное действующее вещество, неинкапсулированное действующее вещество, свободное действующее вещество" (unentrapped, unencapsulated or free active substance) - действующее вещество, находящееся вне липосомы. В рамках настоящего Руководства свободная концентрация равнозначна концентрации несвязанного действующего вещества независимо от того, связано ли действующее вещество с плазменными или иными тканевыми белками;
"общая концентрация действующего вещества" (total concentration of active substance) - суммарная концентрация связанного и несвязанного с липосомами действующего вещества;
"связанное действующее вещество, инкапсулированное действующее вещество" (entrapped or encapsulated active substance) - действующее вещество, находящееся внутри липосомы и отделенное от биологической матрицы одной или более липидной мембраной;
"текучесть мембраны" (membrane fluidity) - способность большей части липидов и белков, входящих в состав мембраны, диффундировать в пределах ее липидного бислоя.
III. Общие требования к качеству липосомальных
лекарственных препаратов
10. Критичные показатели качества липосомальных лекарственных препаратов могут оказывать значимое влияние на фармакокинетические и фармакодинамические свойства in vivo по следующим причинам:
а) скорость высвобождения действующего вещества из липосом может влиять на фармакокинетику и фармакодинамику, а также на профиль безопасности и эффективности липосомального лекарственного препарата;
б) связанное действующее вещество может не обладать биологической доступностью и может быть защищено от деградации, а также от метаболизма при нахождении в липосоме;
в) фармакокинетика связанного действующего вещества может контролироваться фармакокинетикой носителя (то есть липосом), на которую в свою очередь влияют:
физико-химические свойства липосом;
физико-химическое состояние связанного действующего вещества;
взаимодействие, которое возникает между компонентами липосомы и биологической средой или которое определяется ими;
г) состав препарата может влиять на поглощение и распределение действующего вещества в тканях.
11. Перед началом доклинической и клинической разработки липосомального лекарственного препарата следует подтвердить фармацевтическую сопоставимость между воспроизведенным и референтным липосомальными лекарственными препаратами. Вследствие сложности состава липосомальных лекарственных препаратов изолированное подтверждение фармацевтической сопоставимости с референтным препаратом не может заменить доклинические и (или) клинические данные, но может служить обоснованием сокращения объема таких исследований. Объем и сложность доклинических и клинических исследований должны определяться результатами исследований сопоставимости на каждой стадии.
1. Установление характеристик качества
12. Критичным для обеспечения качества липосомального лекарственного препарата является правильное определение его значимых физико-химических свойств. При подготовке регистрационных досье липосомальных лекарственных препаратов всех видов необходимо изучить следующие параметры:
а) критический анализ липидных компонентов (описание, источник и установление характеристик, производство, количественное определение, профиль примесей, изомеры и характеристики стабильности);
б) качество, чистота и стабильность прочих критичных вспомогательных веществ;
в) идентификация и контроль ключевых промежуточных продуктов процесса производства;
г) соответствие отношения "действующее вещество - липидные компоненты" на значимых стадиях производства допустимому диапазону (в целях обеспечения постоянства функциональных характеристик препарата);
д) морфология, средний размер и распределение липосом по размеру, наличие агрегации;
е) доля связанного действующего вещества (отношение свободного к связанному);
ж) стабильность действующего вещества, липидов и функциональных вспомогательных веществ в готовом препарате, включая количественное определение критичных продуктов деградации (например, лизофосфатидилхолина, окисленных (гидролизованных) фрагментов);
з) скорость высвобождения действующего вещества из липосомы in vitro в физиологических (клинически значимых) средах.
13. Следует разработать надежные и обладающие дискриминирующей способностью валидированные методы оценки высвобождения in vitro в целях:
проведения мониторинга имитации высвобождения действующего вещества из липосом в физиологических (клинически значимых) средах. При наличии обоснований допустимо испытание на утечку (leakage test) in vitro в релевантной среде в различных условиях (например, в диапазоне температур и значений рН);
проведения мониторинга стабильности при хранении (он должен быть достаточно чувствительным, чтобы обеспечивать постоянство серий);
исследования стабильности в предполагаемых условиях применения;
исследования устойчивости процесса восстановления и (или) приготовления в аптеке.
14. Качество и чистота исходных липидных материалов - основополагающий фактор качества лекарственного препарата, в связи с этим особенно важным является надлежащее установление характеристик и составление спецификаций на исходный липидный материал. Необходимо должным образом проанализировать характеристики, определяющие функциональность, в соответствии со статьей "Функциональные характеристики вспомогательных веществ" Фармакопеи Союза, а при отсутствии в ней - с соответствующими статьями фармакопей государств-членов. Объем представляемых сведений в составе регистрационного досье зависит от сложности вспомогательных веществ. Использование нескольких источников (например, животных, растительных, синтетических) или поставщиков липидных компонентов потребует дополнительного проведения исследований по установлению их характеристик и сопоставимости.
15. В зависимости от конкретной функции липосом (например, модификации распределения действующего вещества путем инкапсуляции в целях улучшения профиля безопасности или модификации фармакокинетики липосом посредством пэгилирования) в регистрационном досье также указываются следующие дополнительные параметры:
а) поддержание целостности готовой липосомальной формы в плазме;
б) характеристика процесса фазового перехода липидного бислоя (например, температура и энтальпия переходов);
в) определение поверхностного заряда липосом;
г) значение рН внутренней камеры липосом, наполняемых по градиенту рН;
д) установление характеристик физического состояния действующего вещества внутри липосомы (например, в случае доксорубицина - образование осадка) (если значимо);
е) распределение действующего вещества внутри липосомы (например, на поверхности, в бислое, внутренней среде);
ж) в отношении конъюгированных (например, пэгилированных) липосомальных лекарственных препаратов:
з) качество и чистота пэгилированного исходного материала, являющиеся основополагающими факторами качества лекарственного препарата;
и) химические данные о достижении конъюгации (например, ПЭГ-липиды или аналогичные конструкции с полиэтиленгликолем или без него);
к) молекулярная масса конъюгированного липида и распределение по размеру (дисперсность);
л) расположение полиэтиленгликоля на поверхности;
м) стабильность конъюгата.
16. Необходимо определить перечень испытаний, которым липосомальный лекарственный препарат планируется подвергать рутинно. Он должен основываться на параметрах, использованных для характеристики препарата в соответствии с пунктами 11, 12 и 14 настоящего Руководства.
2. Установление фармацевтической сопоставимости
17. Качественный и количественный состав разрабатываемого липосомального лекарственного препарата должен быть идентичным составу референтного липосомального лекарственного препарата или практически совпадать с ним.
18. Как правило, заявитель липосомального лекарственного препарата, разрабатываемого по аналогии с референтным препаратом, не имеет доступа к сведениям о процессе производства референтного липосомального лекарственного препарата. В целях получения доказательств того, что характеристики разрабатываемого и референтного липосомального лекарственного препарата сопоставимы, необходимо выполнить расширенную программу испытаний, с применением современных методов параллельного установления характеристик обоих липосомальных лекарственных препаратов. В эту программу необходимо включить все значимые испытания, описанные в подразделе 1 настоящего раздела, подходящие для правильной характеристики воспроизведенного липосомального лекарственного препарата и липосомального лекарственного препарата сравнения, в особенности в отношении характеристики их функциональных свойств in vivo.
19. Необходимо проанализировать значимость выбранных испытаний для подтверждения эквивалентности функциональных характеристик липосомального лекарственного препарата in vivo. Все различия между препаратами, обнаруженные в исследованиях сопоставимости, необходимо принять во внимание, детально оценить и обосновать с точки зрения влияния на безопасность и (или) эффективность.
20. Помимо исследований по установлению характеристик, проведенных в нормальных условиях, в целях сравнения физической и химической деградации необходимо провести сравнительные стресс-испытания обоих препаратов.
21. Анализ всех серий референтного лекарственного препарата в исследованиях по установлению характеристик необходимо провести в пределах их срока годности, их хранение перед анализом следует осуществлять в рекомендуемых условиях.
3. Фармацевтическая разработка липосомального
лекарственного препарата
22. В целях обеспечения производства липосомального лекарственного препарата с приемлемым качеством на постоянной основе необходимо располагать хорошо описанным процессом производства с удовлетворительным контролем такого процесса. Вместе с тем известно, что небольшие изменения в липосомальных лекарственных препаратах могут существенно повлиять на их функциональные характеристики. Подходы к определению влияния любого изменения процесса производства зависят от конкретного процесса производства, препарата, объема знаний и опыта, полученного производителем ранее в отношении подобного процесса, а также от имеющихся в наличии данных по разработке липосомального лекарственного препарата. При изменении процесса производства на этапе разработки, а также на пострегистрационном этапе (например, при масштабировании) необходимо провести сравнительные исследования.
23. Если результаты физико-химических испытаний свидетельствуют об изменении свойств липосомального лекарственного препарата, могут потребоваться исследования in vivo, направленные на подтверждение того, что никакие изменения не повлияли на профиль безопасности и эффективности.
24. Заявителю рекомендуется принять во внимание базовые принципы, изложенные в разделе 1.4 главы 9.1 Правил проведения исследований биологических лекарственных средств Евразийского экономического союза, утвержденных Решением Совета Евразийской экономической комиссии от 3 ноября 2016 г. N 89.
IV. Стратегия доклинических и клинических исследований
1. Общие положения
25. Документация, необходимая для регистрации липосомального лекарственного препарата, разрабатываемого в качестве аналога референтного липосомального лекарственного препарата, должна быть достаточно детализирована, чтобы гарантировать обоснованность заключения об эквивалентной безопасности и эффективности по отношению к референтному лекарственному препарату. Доклинические исследования проводятся до начала клинических исследований и, как правило, предусматривают сравнительное изучение фармакокинетики (включая распределение в тканях), токсикологию и фармакодинамику. Однако сложность конкретного липосомального лекарственного препарата определяет возможность уменьшения объема сравнительных доклинических исследований, и, при наличии такой возможности, исключение конкретных исследований определяется в индивидуальном порядке.
26. При всесторонней оценке нового липосомального лекарственного препарата данные, полученные по результатам фармацевтических, доклинических и клинических исследований, следует анализировать как единое целое. Например, если по результатам доклинических исследований выявлены какие-либо значимые отличия липосомального лекарственного препарата, разрабатываемого в качестве аналога референтного лекарственного препарата, рекомендуется провести критическую переоценку физико-химических характеристик такого липосомального лекарственного препарата, чтобы до начала клинических исследований выяснить возможные причины таких отличий. Различия в данных, полученных при изучении аналогичности воспроизведенного липосомального лекарственного препарата референтному липосомальному лекарственному препарату, позволяют сделать заключение об отсутствии сопоставимости таких липосомальных лекарственных препаратов и являются основанием для формулировки соответствующего заключения уполномоченными органами государств-членов при рассмотрении заявления о регистрации такого лекарственного препарата.
27. Если действующее вещество вводится в виде липосомального лекарственного препарата, обнаруживаются существенные изменения фармакокинетических характеристик. Так объем распределения и клиренс могут снижаться, а период полувыведения увеличиваться. Клиренс липосомального действующего вещества зависит от:
клиренса самого липосомального носителя;
скорости высвобождения связанного действующего вещества из липосомального носителя;
клиренса и метаболизма несвязанного действующего вещества после его высвобождения.
Скорость и место высвобождения действующего вещества in vivo - ключевой параметр, влияющий на токсичность и эффективность.
Следовательно, фармакокинетику воспроизведенного липосомального лекарственного препарата следует всегда сравнивать с фармакокинетикой референтного липосомального лекарственного препарата. Применимы лишь некоторые аспекты традиционного подхода к изучению биоэквивалентности, а в некоторых случаях необходимо вводить дополнительные требования, определяемые в индивидуальном порядке.
28. В сравнительных фармакокинетических исследованиях необходимо подтвердить не только аналогичность общей экспозиции несвязанного и связанного в липосому действующего вещества (в пунктах 39 и 45 настоящего Руководства указаны аналиты, подлежащие определению в доклинических и клинических исследованиях), но и с их помощью них необходимо также подтвердить аналогичность параметров распределения и клиренса.
2. Методы анализа
29. Помимо традиционных методов определения общего содержания действующего вещества и его метаболитов в крови (плазме) и тканях, в целях сравнения с референтным липосомальным лекарственным препаратом потребуется разработать и валидировать аналитические методы количественного определения связанного и несвязанного действующего вещества в крови (плазме) и несвязанного действующего вещества в тканях.
30. Раздельное количественное определение несвязанного и связанного действующего вещества обусловливает необходимость использования методологий разделения, которые в целях проверки их надежности требуют особого внимания. В каждом образце крови (плазмы) в целях независимой проверки надежности методологии разделения необходимо определить общее содержание действующего вещества без разделения его на связанное и несвязанное. Несмотря на выполнимость количественного определения несвязанного, связанного и суммарного действующего вещества в крови (плазме), признается, что процесс обработки тканей, вероятнее всего, приведет к разрушению липосом.
31. При определении содержания несвязанного действующего вещества в тканях следует особо тщательно подойти к выделению несвязанного действующего вещества до процедур обработки тканей, которые, вероятнее всего, приведут к разрушению липосом. В ходе разработки метода необходимо уделить особое внимание влиянию всех процедур пробоподготовки, прибегая к методологиям верификации пригодности и интерпретируемости всех биоаналитических результатов.
32. Необходимо описать методы анализа, используемые для количественного определения действующего вещества (суммарного, несвязанного и связанного) и метаболитов в плазме и тканях, и их валидацию. Необходимо указать нижние пределы количественного определения и извлечения действующего вещества из плазмы, тканей и, если применимо, отдельных интересующих тканей (например, опухолевых).
V. Доклинические исследования липосомальных
лекарственных препаратов
1. Доклинические фармакодинамические исследования
липосомальных лекарственных препаратов
33. Доклинические фармакодинамические исследования липосомальных лекарственных препаратов должны предусматривать:
а) разработку испытаний in vitro, способных охарактеризовать любое взаимодействие между липосомами и клетками-мишенями или иными клетками, взаимодействие с которыми токсикологически значимо (по возможности). При этом, несмотря на наличие возможности охарактеризовать фармакодинамический профиль липосомального лекарственного препарата только с помощью испытаний in vitro, объем данных, получаемый в таких испытаниях, ограничен и высока вероятность того, что потребуется проведение исследований in vivo;
б) подтверждение сопоставимости фармакодинамического ответа с использованием надлежащих in vivo моделей и при разных выбранных дозах с учетом чувствительности модели.
2. Доклинические фармакокинетические исследования
липосомальных лекарственных препаратов
34. Некоторые фармакокинетические параметры липосомальных лекарственных препаратов с позиции их функциональных характеристик у человека можно спрогнозировать на животных и, если применимо, на моделях на основе клеток. Однако выбор релевантных видов животных и моделей в целях изучения высвобождения действующего вещества из липосом in vivo следует обосновать, уделяя особое внимание кумуляции и удержанию в органах-мишенях, фармакокинетике и распределению.
35. Помимо системной экспозиции, следует подтвердить аналогичность распределения и элиминации. Эти исследования дают опорное подтверждение сопоставимости фармакокинетики липосомальных лекарственных препаратов, поскольку невозможно получить полную картину распределения у человека, опираясь только на данные по крови (плазме). По этой причине исследования необходимо проводить в соответствии с Правилами надлежащей лабораторной практики Евразийского экономического союза в сфере обращения лекарственных средств, утвержденными Решением Совета Евразийской экономической комиссии от 3 ноября 2016 г. N 81, на видах животных, подходящих с точки зрения фармакологии и безопасности препарата. Воспроизведенный липосомальный лекарственный препарат необходимо произвести с помощью полномасштабного (промышленного) процесса производства, в доклинических исследованиях оптимально использовать ту же серию липосомального лекарственного препарата, которая будет в последующем изучена в опорных клинических исследованиях.
36. Необходимо тщательно продумать выбор временных точек и продолжительности взятия образцов, чтобы можно было точно количественно определить динамику изменения концентрации несвязанного и суммарного содержания действующего вещества и метаболита в тканях, уравновешивая необходимость количественного определения раннего высвобождения действующего вещества из липосом (например, в течение первых 15 минут) с необходимостью изучения персистенции действующего вещества в определенных тканях. Если исходя из возможностей существующих аналитических методов свободную концентрацию измерить невозможно, необходимо предпринять попытки сравнения концентрации метаболитов в органах-мишенях. Поскольку такие исследования предполагают разрушение образцов при отборе, число животных, подлежащих включению в исследование, зависит от числа временных точек отбора образцов, вариабельности тканевого распределения действующего вещества между особями и вариабельности, обусловленной проведением эксперимента (иссечения ткани, взвешивания, гомогенизации и пробоподготовке, а также биоаналитических источников вариабельности). Тщательный выбор времени отбора образцов повысит прецизионность получаемых результатов. Во избежание неудачных опорных исследований и неинтерпретируемости их результатов, в целях установления правильных доз, необходимой стратегии взятия образцов и числа используемых животных рекомендуется проводить пилотные исследования. Для анализа следует выбрать ткани, позволяющие определить безопасность и эффективность липосомального лекарственного препарата, а также ткани, обеспечивающие значимую часть процессинга (элиминации) липосом. В связи с ограниченностью опыта проведения таких исследований невозможно определить конкретные критерии сопоставимости распределения действующего вещества в тканях.
37. Рекомендуется использовать повторные (репликативные) дизайны исследований, в которых репликативно вводится по меньшей мере только референтный лекарственный препарат, иначе любые различия между воспроизведенным препаратом и референтным лекарственными препаратами не будут поддаваться интерпретации. С целью снижения вариабельности результатов следует предусмотреть использование правильно выбранного внутреннего стандарта. Полученные данные следует представлять различными способами, в том числе приводя различия в фармакокинетических параметрах между лекарственными препаратами, а также визуальные сравнения профилей "концентрация - время" для каждого вида ткани и для каждого аналита. Все оценки и способы представления данных необходимо сопроводить вычислением неопределенности (например, доверительными интервалами). Необходимо проанализировать клинические последствия всех выявленных различий в распределении действующего вещества в тканях между воспроизведенным препаратом и референтным препаратом.
3. Доза липосомальных лекарственных препаратов, подлежащая
изучению в доклинических исследованиях
38. Для обоснования сопоставимой фармакокинетики могут потребоваться исследования с однократным и многократным введением нескольких уровней доз лекарственных препаратов. При выборе доз лекарственных препаратов следует исходить из концентрации действующего вещества липосомального лекарственного препарата в крови у человека при введении терапевтических доз такого липосомального лекарственного препарата. С целью установления правильной дозы лекарственных препаратов рекомендуется использовать аллометрические уравнения и физиологически обоснованные фармакокинетические модели.
4. Аналиты в биологических жидкостях, подлежащие
определению в доклинических исследованиях
39. Следует по возможности изучить кинетику (включая тканевое распределение и экскрецию) как несвязанного действующего вещества, так и связанного.
5. Токсикологические исследования липосомальных
лекарственных препаратов
40. Проведение токсикологического исследования липосомальных лекарственных препаратов может не потребоваться. Вместе с тем в зависимости от результатов изучения фармацевтической сопоставимости и характера токсичности липосомального лекарственного препарата с целью обоснования эквивалентности в контексте известной токсичности могут потребоваться исследования функций органов-мишеней (например, в случае подозрения на кардиотоксичность может оказаться целесообразной оценка функции сердца путем измерения конечно-диастолического давления в левом желудочке у крыс).
41. В целях оценки потенциала развития нежелательных явлений необходимо предусмотреть использование испытания in vitro и исследования in vivo на иммунную реактогенность, такие как определение активации комплемента и (или) макрофагов (базофилов) и испытание на обусловленную активацией комплемента псевдоаллергическую реакцию на чувствительных моделях животных.
VI. Клинические исследования липосомальных
лекарственных препаратов
1. Сравнительные фармакокинетические исследования
липосомальных лекарственных препаратов
Доза липосомальных лекарственных препаратов, подлежащая
изучению в клинических исследованиях
42. Фармакокинетические свойства нередко зависят от дозы липосомального лекарственного препарата, поэтому при отсутствии подтверждения линейности воспроизведенный и референтный липосомальные лекарственные препараты следует сравнивать в рекомендуемом диапазоне доз. При отсутствии научных данных заявляемую линейность необходимо подтвердить для связанного, несвязанного и суммарного действующего вещества.
43. При нелинейности достаточно подтверждения биоэквивалентности для наибольшей и наименьшей доз липосомального лекарственного препарата, даже если разные дозы применяются при разных показаниях. В таких случаях дополнительные клинические исследования не проводятся. В тех случаях, когда из этических или иных соображений определенные дозы невозможно изучить в исследованиях биоэквивалентности, оценка терапевтической эквивалентности по каждому показанию к применению требует индивидуального подхода.
Вопросы дизайна исследований липосомальных
лекарственных препаратов
44. В случае если здоровые добровольцы плохо переносят действующее вещество, фармакокинетическое исследование можно провести с участием пациентов. Если исследование с однократным введением доз невыполнимо, допускается проведение фармакокинетических исследований с повторным (многократным) введением доз.
Аналиты в биологических жидкостях, подлежащие определению
в клинических исследованиях после введения липосомальных
лекарственных препаратов
45. Валидированный биоаналитический метод должен предусматривать надежное количественное определение суммарного, связанного и несвязанного действующего вещества. Поскольку метаболизм действующего вещества начинается только после его высвобождения из липосом, количественное определение по меньшей мере одного метаболита, независимо от его фармакологической активности, может способствовать оценке и сравнению скорости высвобождения действующего вещества из липосомального лекарственного препарата. При наличии нескольких метаболитов при выборе одного из них следует руководствоваться кинетическими параметрами. Если один или несколько метаболитов обладают значимой клинической активностью, может потребоваться также сравнение их кинетики.
Фармакокинетические параметры, подлежащие определению
и документированию в клинических исследованиях
46. Фармакокинетические параметры суммарного, связанного и несвязанного действующего вещества, подлежащие определению и документированию, должны позволять провести сравнение скорости, с которой действующее вещество высвобождается из липосом, поскольку она будет определять начало и продолжительность терапевтического эффекта. Вместе с тем такие стандартные фармакокинетические параметры, как AUC и Cmax, могут недостаточно характеризовать скорость высвобождения в тканях-мишенях. В связи с этим с целью описания других фармакокинетических процессов, в том числе распределения и элиминации, в дополнение к данным о скорости и степени высвобождения необходимо представить данные об изучении дополнительных фармакокинетических параметров. Если это значимо, необходимо сравнить скорость и степень экскреции действующего вещества в мочу.
47. В целях обеспечения сопоставимости раннего клиренса ретикулоэндотелиальной системой необходимо предусмотреть ранние временные точки отбора образцов во время и немедленно после завершения инфузии препарата.
48. Если скорость элиминации несвязанного и связанного действующего вещества различается, что характерно для липосом с более длительным высвобождением действующего вещества, необходимо предоставить данные о дополнительных фармакокинетических параметрах, таких как клиренс, объем распределения, терминальный период полувыведения и частичные AUC (например, 0 - 24 ч, 24 - 48 ч и т.д.). Эти параметры необходимо оценить описательно. Это может потребовать дальнейшей характеристики целостности липосом и их захвата периферическими тканями и (или) ретикулоэндотелиальной системой. Кроме того, можно предусмотреть дополнительные описательные параметры, например межкамерный клиренс и объем периферической и центральной камер. Рекомендуется определять динамику отношения концентрации несвязанного и связанного действующего вещества.
Критерии приемлемости показателей эквивалентности
для липосомальных лекарственных препаратов
49. Необходимо подтвердить, что концентрации суммарного, связанного и несвязанного вещества аналогичны. Доверительные интервалы для отношений Cmax, 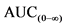 и AUC(0-t) должны, как правило, укладываться в диапазон 80,00 - 125,00%. В отдельных случаях к дополнительным параметрам относят частичные AUC или критерии приемлемости для фармакокинетических параметров метаболита.
и AUC(0-t) должны, как правило, укладываться в диапазон 80,00 - 125,00%. В отдельных случаях к дополнительным параметрам относят частичные AUC или критерии приемлемости для фармакокинетических параметров метаболита.
2. Исследования со сравнительной оценкой эффективности
липосомальных лекарственных препаратов
50. Необходимость проведения клинического исследования (исследований) эффективности в дополнение к обязательным клиническим фармакокинетическим исследованиям, как правило, определяется в индивидуальном порядке, в зависимости от способности доклинических моделей и клинических фармакокинетических данных обнаруживать различия между референтным липосомальным лекарственным препаратом и препаратом, разрабатываемым с ним по аналогии, а также сложности состава и строения липосом.
51. Если референтный и разрабатываемый лекарственные препараты различаются по качественному составу, высока вероятность того, что потребуются дополнительные исследования терапевтической эквивалентности (например, в случае соединения полимеров с липидами с помощью различных способов связывания).
52. Вместе с тем вследствие относительной нечувствительности клинических исследований к обнаружению различий, обусловленных различиями в составах и способе производства липосомальных лекарственных препаратов, этот подход не является предпочтительным. В связи с этим при разработке липосомального лекарственного препарата как аналога референтного липосомального лекарственного препарата необходимо приложить все возможные усилия для подтверждения эквивалентности фармацевтического качества таких препаратов и аналогичности в ходе доклинических фармакокинетических и фармакодинамических, а также клинических фармакокинетических исследованиях. В случае если в данных, полученных в процессе исследований, проводимых с целью обоснования аналогичности, обнаружены различия между референтным и воспроизведенным липосомальными лекарственными препаратами, то такие липосомальные лекарственные препараты рассматриваются как не являющиеся аналогичными, а представленные данные являются основанием для замечаний уполномоченных органов (экспертных организаций) государств-членов в отношении выполненных исследований.
3. Исследования с оценкой безопасности липосомальных
лекарственных препаратов
53. Острые инфузионные реакции на липосомальные лекарственные препараты относятся к распространенным нежелательным реакциям. При этом частота таких нежелательных реакций будет сопоставима, если воспроизведенные липосомальные лекарственные препараты не различаются по качественному составу (в состав лекарственных препаратов могут входить различные вспомогательные вещества) или процессу производства. С целью предотвращения несовпадения профиля безопасности необходимо, чтобы качественный и количественный состав воспроизведенного препарата был идентичен или максимально совпадал с референтным лекарственным препаратом. Вместе с тем в целях минимизации частоты развития острых инфузионных реакций требуется проводить испытания in vitro и исследования in vivo на иммунную реактогенность, рассмотренные в пунктах 40 и 41 настоящего Руководства. При наличии признаков того, что новый липосомальный лекарственный препарат может приводить к повышенному риску развития таких реакций, следует проанализировать разработку лекарственного препарата с целью выяснения их причин. Более того, необходимо тщательно оценить инфузионные реакции в рамках исследований биоэквивалентности, а также проанализировать сведения и данные по разработке воспроизведенного липосомального лекарственного препарата.
54. Как правило, до регистрации воспроизведенного липосомального лекарственного препарата не требуется проведение полномасштабных клинических исследований. Клиническую безопасность воспроизведенных липосомальных лекарственных препаратов следует оценивать в процессе их обращения в соответствии с актами, входящими в право Евразийского экономического союза, включая Правила надлежащей практики фармаконадзора Евразийского экономического союза, утвержденные Решением Совета Евразийской экономической комиссии от 3 ноября 2016 г. N 87.