Рекомендация Коллегии Евразийской экономической комиссии от 11.11.2025 N 30
КОЛЛЕГИЯ ЕВРАЗИЙСКОЙ ЭКОНОМИЧЕСКОЙ КОМИССИИ
РЕКОМЕНДАЦИЯ
от 11 ноября 2025 г. N 30
О РУКОВОДСТВЕ
ПО ФАРМАЦЕВТИЧЕСКОЙ РАЗРАБОТКЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ
Коллегия Евразийской экономической комиссии в соответствии с пунктом 3 статьи 3, пунктом 2 статьи 4 и пунктом 7 статьи 7 Соглашения о единых принципах и правилах обращения лекарственных средств в рамках Евразийского экономического союза от 23 декабря 2014 года, в целях установления общих подходов к фармацевтической разработке лекарственных средств
рекомендует государствам - членам Евразийского экономического союза по истечении 30 календарных дней с даты опубликования настоящей Рекомендации на официальном сайте Евразийского экономического союза при фармацевтической разработке лекарственных средств принимать во внимание Руководство согласно приложению.
Председатель Коллегии
Евразийской экономической комиссии
Б.САГИНТАЕВ
Приложение
к Рекомендации Коллегии
Евразийской экономической комиссии
от 11 ноября 2025 г. N 30
РУКОВОДСТВО
ПО ФАРМАЦЕВТИЧЕСКОЙ РАЗРАБОТКЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ
I. Общие положения
1. Настоящее Руководство содержит указания по составлению раздела 3.2.P.2 "Фармацевтическая разработка" регистрационного досье лекарственного препарата (далее - регистрационное досье) в формате общего технического документа (ОТД). Раздел "Фармацевтическая разработка" регистрационного досье дает возможность заявителю представить имеющиеся у него данные и сведения, полученные при применении научных подходов и при управлении рисками для качества во время разработки лекарственного препарата и в процессе его производства. Указанный раздел является частью регистрационного досье лекарственного препарата, подаваемого на регистрацию, и впоследствии может обновляться держателем регистрационного удостоверения на основании новых данных и сведений, полученных в течение жизненного цикла лекарственного препарата. Раздел "Фармацевтическая разработка" регистрационного досье позволяет обеспечить всестороннее понимание характеристик лекарственного препарата и процесса его производства экспертами уполномоченных органов (экспертных организаций) и инспекторами фармацевтических инспекторатов государств - членов Евразийского экономического союза (далее - государства-члены, Союз). Настоящее Руководство также определяет те области фармацевтической разработки, в которых подтверждение заявителем глубокого понимания и имеющихся научных знаний в фармацевтической области и знаний о производстве лекарственного препарата позволит уполномоченным органам (экспертным организациям) государств-членов применять более гибкие подходы к оценке регистрационного досье. Степень гибкости подходов, применяемых уполномоченными органами (экспертными организациями) государств-членов, будет определяться уровнем представленных научных знаний.
2. Настоящее Руководство содержит описание принципов проектирования качества (QbD) лекарственного препарата и не направлено на установление новых стандартов или введение новых требований. В настоящем Руководстве показано, как заявитель может на практике применить концепции и инструменты фармацевтической разработки (например, проектное поле) в отношении различных лекарственных форм.
Если фармацевтический производитель при разработке лекарственного препарата решает применить к фармацевтической разработке проектирование качества и управление рисками для качества, предусмотренные частью III Правил надлежащей производственной практики Евразийского экономического союза, утвержденных Решением Совета Евразийской экономической комиссии от 3 ноября 2016 г. N 77 (далее - Правила производственной практики), то он вправе использовать эти дополнительные возможности для дальнейшего получения научных знаний и применения риск-ориентированных подходов к управлению качеством лекарственного препарата.
II. Сфера применения
3. Настоящее Руководство не применяется к составлению досье лекарственных препаратов, находящихся на разных фазах клинической разработки. Вместе с тем положения настоящего Руководства допускается учитывать на этих фазах. Настоящее Руководство может также применяться к разным видам разрабатываемых лекарственных препаратов. В соответствии с пунктом 26 Правил регистрации и экспертизы лекарственных средств для медицинского применения, утвержденных Решением Совета Евразийской экономической комиссии от 3 ноября 2016 г. N 78, заявители вправе дополнительно проконсультироваться с соответствующими уполномоченными органами (экспертными организациями) государства-члена по вопросам применимости настоящего Руководства в отношении конкретного вида разрабатываемого лекарственного препарата.
III. Определения
4. Для целей настоящего Руководства используются понятия, которые означают следующее:
"выпуск в реальном времени" - способность оценивать и обеспечивать качество промежуточной продукции и (или) лекарственного препарата на основании данных о технологическом процессе, которые, как правило, включают в себя надежную комбинацию измеренных характеристик лекарственного препарата (материалов) и видов контроля в процессе производства лекарственного препарата;
"доказанный приемлемый диапазон" - охарактеризованный диапазон параметра технологического процесса, работа в пределах которого (при неизменности остальных параметров) будет приводить к получению материала, соответствующего релевантным критериям качества;
"качество лекарственного средства" - пригодность активной фармацевтической субстанции или лекарственного препарата для целевого назначения, в том числе по таким показателям, как идентификация, количественное определение и примеси;
"планирование эксперимента (дизайн эксперимента)" - структурированный, организованный метод определения зависимости между факторами, влияющими на процесс, и результатом такого процесса;
"проектирование качества (QbD)" - систематический подход к разработке лекарственных препаратов, основанный на строгих научных принципах и управлении рисками для качества, начинающийся с предварительного определения целей и делающий акцент на понимании предназначения лекарственного препарата и технологического процесса, а также внутрипроизводственный контроль;
"проектное поле" - многомерная комбинация и взаимодействие входных переменных (например, характеристик материалов) и параметров технологического процесса, подтвердившие способность обеспечивать качество продукта;
"процессно-аналитическая технология (PAT)" - система проектирования, анализа и контроля процесса производства посредством своевременных измерений (во время обработки) критических показателей качества и функциональных характеристик сырья и внутрипроизводственных материалов, находящихся в процессе обработки, а также технологических процессов в целях обеспечения качества продукта (в том числе лекарственного препарата);
"робастность (устойчивость) процесса" - способность технологического процесса выдерживать вариабельность материалов, а также изменений процесса и оборудования без негативного влияния на качество продукта (в том числе лекарственного препарата);
"стратегия контроля" - комплекс средств контроля, спланированный и составленный на основании известных на данном этапе характеристик и свойств лекарственного препарата и технологического процесса и обеспечивающий надлежащее осуществление этого технологического процесса и качество лекарственного препарата;
"целевой профиль качества лекарственного препарата (ЦПКП)" - планируемая совокупность показателей качества лекарственного препарата, которую потенциально следует достигнуть, чтобы обеспечить заданное качество (с учетом безопасности и эффективности лекарственного препарата).
Понятия "жизненный цикл продукции", "жизненный цикл лекарственного препарата", "критический показатель качества", "критический параметр процесса" и "непрерывная верификация процесса" используемые в настоящем Руководстве, применяются в значениях, определенных Правилами производственной практики.
IV. Фармацевтическая разработка
5. Целью фармацевтической разработки являются создание качественного лекарственного препарата и процесса его производства, которые будут постоянно обеспечивать заданные характеристики лекарственного препарата. Сведения и знания, получаемые по результатам исследований по фармацевтической разработке, и производственный опыт позволяют достичь понимания и обосновать установление проектного поля, спецификаций и подходов к производственному контролю.
6. Сведения, полученные по результатам исследований по фармацевтической разработке, могут стать основой управления рисками для качества. Поскольку невозможно гарантировать качество лекарственного препарата только путем проведения тестирования этого лекарственного препарата, его качество закладывается в препарат при проектировании (дизайне). Внесение изменений в состав на серию (производственную рецептуру) и в процессы производства во время фармацевтической разработки и управления жизненным циклом лекарственного препарата следует рассматривать как возможность для получения дополнительных знаний и дальнейшего обоснования создания проектного поля. Целесообразно также включить в отчет о фармацевтической разработке релевантные данные, полученные из экспериментов, давших неожиданные результаты. Проектное поле предлагается заявителем в составе регистрационного досье и подлежит экспертной оценке при регистрации лекарственного препарата или внесении изменений в его регистрационное досье.
Работа фармацевтического производителя в пределах проектного поля не рассматривается в качестве изменения в регистрационное досье. Выход за пределы проектного поля рассматривается в качестве изменения и, как правило, влечет за собой внесение изменений в регистрационное досье.
7. В раздел "Фармацевтическая разработка" регистрационного досье включаются сведения, подтверждающие, что выбранный вид лекарственной формы, а также предлагаемые состав и технология подходят для целевого назначения лекарственного препарата. В каждую часть этого раздела следует включать сведения, чтобы позволить надлежащим образом представить разработку лекарственного препарата и его технологический процесс, обобщающие таблицы и схемы (если они повышают уровень понимания и облегчают экспертизу).
8. По меньшей мере следует определить те аспекты фармацевтических субстанций, вспомогательных веществ, систем упаковки (укупорки) и технологических процессов, которые критичны для качества лекарственного препарата, и обосновать стратегии контроля. Критичные показатели состава и параметры технологического процесса, как правило, выявляются при помощи оценки того, насколько их вариабельность может влиять на качество лекарственного препарата.
9. Заявитель вправе принять решение о проведении исследований по фармацевтической разработке, по результатам которых можно получить более обширные сведения о функциональных характеристиках лекарственного препарата в более широком диапазоне показателей качества материалов, вариантов обработки и параметров технологического процесса. Включение таких дополнительных сведений в этот раздел дает возможность продемонстрировать большую степень понимания характеристик материалов, технологических процессов и их контроля. Такое научное понимание облегчает установление расширенного проектного поля. В таких случаях существует возможность для разработки более гибких подходов к регулированию обращения лекарственных средств, например, чтобы способствовать:
а) принятию уполномоченными органами (экспертными организациями) государств-членов риск-ориентированных решений;
б) совершенствованию процесса производства в пределах одобренного проектного поля, описанного в регистрационном досье, без проведения экспертизы уполномоченным органом (экспертной организацией) государства-члена при внесении изменений в регистрационное досье;
в) снижению количества заявлений о внесении изменений в регистрационное досье;
г) внедрению внутрипроизводственного контроля качества в реальном времени, приводящего к сокращению испытаний выпускающего контроля готового продукта (в том числе лекарственного препарата).
10. При выборе подхода, указанного в пункте 9 настоящего Руководства, заявителю следует представить более детальные сведения о функциональных характеристиках лекарственного препарата в диапазоне показателей качества материалов, возможностях технологического процесса и его параметрах. Такие сведения можно получить с применением, например, планирования эксперимента, процессно-аналитической технологии (PAT) и (или) предварительных знаний. Соответствующее использование принципов управления рисками для качества может оказаться полезным при выборе приоритетов при принятии решений о необходимости проведения дополнительных исследований по фармацевтической разработке.
11. Планирование и проведение исследований по фармацевтической разработке выполняются исходя из планируемой научной цели. Уровень полученных знаний (а не объем данных) является основой для формирования научно обоснованных сведений, включаемых в материалы регистрационного досье, и проведения его экспертизы уполномоченным органом (экспертной организацией) государства-члена.
1. Компоненты лекарственного препарата
Активная фармацевтическая субстанция
12. Для активной фармацевтической субстанции следует определить и изучить:
а) физико-химические и биологические свойства активной фармацевтической субстанции, способные влиять на функциональные характеристики лекарственного препарата;
б) пригодность активной фармацевтической субстанции для технологического процесса;
в) свойства, которые были целенаправленно встроены в активную фармацевтическую субстанцию (например, свойства твердой фазы).
13. К физико-химическим и биологическим свойствам, которые необходимо будет изучить, относятся растворимость, содержание воды, размер частиц, кристаллические свойства, биологическая активность и проникающая способность.
14. Свойства, указанные в подпунктах "а" - "в" пункта 12 настоящего Руководства, могут быть взаимосвязаны и их следует рассматривать в совокупности.
15. Чтобы оценить потенциальное влияние физико-химических свойств активной фармацевтической субстанции на поведение лекарственного препарата, следует провести исследования лекарственного препарата. Критерии, при которых следует проводить исследования на лекарственном препарате, описываются в приложении N 1 к Руководству по составлению нормативного документа по качеству лекарственного препарата, утвержденному Решением Евразийской экономической комиссии от 7 сентября 2018 г. N 151 (далее - Руководство по нормативному документу) (например, в схемах решений N 6 и 7). Эти же критерии допускается применять для проектирования качества и разработки спецификаций биологических лекарственных препаратов, предусмотренных главой 6 Правил проведения исследований биологических лекарственных средств Евразийского экономического союза, утвержденных Решением Совета Евразийской экономической комиссии от 3 ноября 2016 г. N 89 (далее - Правила исследований биологических лекарственных средств). Результаты исследований по изучению потенциального влияния свойств активной фармацевтической субстанции на функциональные характеристики лекарственного препарата можно использовать (если применимо) для обоснования спецификации на активную фармацевтическую субстанцию в подразделе 3.2.S.4.5 регистрационного досье.
16. Следует оценить совместимость активной фармацевтической субстанции со вспомогательными веществами, указанными в разделе 3.2.P.1 регистрационного досье. В отношении лекарственных препаратов, содержащих более одной активной фармацевтической субстанции, следует также оценить совместимость этих фармацевтических субстанций друг с другом.
Вспомогательные вещества
17. Следует рассмотреть выбранные вспомогательные вещества, их содержание (концентрацию) и характеристики, способные влиять на функциональные показатели (например, стабильность, биодоступность) или пригодность для технологического процесса лекарственного препарата с точки зрения соответствующей функции каждого вспомогательного вещества. Это применимо ко всем веществам, используемым в производстве лекарственного препарата, независимо от того, попадают ли они в лекарственный препарат (например, технологические добавки). Следует установить совместимость вспомогательных веществ с другими вспомогательными веществами в тех случаях, когда это важно (например, введение двойной системы консервантов). Следует также подтвердить способность вспомогательных веществ (например, антиоксидантов, усилителей проницаемости, дезинтеграторов, средств, контролирующих высвобождение) обеспечивать целевую функциональность и сохранять ее на протяжении всего планируемого срока годности (срока хранения) лекарственного препарата. При необходимости сведения о функциональных характеристиках вспомогательных веществ следует использовать для обоснования выбора вспомогательного вещества и показателей его качества, а также для обоснования спецификации на лекарственный препарат в подразделе 3.2.P.5.6 регистрационного досье.
18. Следует привести ссылки на сведения, обосновывающие безопасность вспомогательных веществ, в подразделе 3.2.P.4.6 регистрационного досье (если применимо).
2. Лекарственный препарат
Разработка лекарственной формы
19. Следует предоставить в регистрационном досье резюме, описывающее разработку состава, в том числе указать те показатели, которые критичны для качества лекарственного препарата с учетом его планируемого применения и пути введения (способа применения). Для выявления потенциально важных для обеспечения качества лекарственного препарата критических или взаимодействующих переменных, следует использовать сведения, полученные во время планирования эксперимента.
20. В резюме следует описать процесс изменения состава, лекарственной формы и формы выпуска лекарственного препарата (дизайн лекарственного препарата) (начиная с первоначальной версии состава лекарственного препарата и заканчивая окончательным составом (производственной рецептурой) лекарственного препарата)). При подготовке резюме следует учитывать выбор компонентов лекарственного препарата (например, исходя из свойств активной фармацевтической субстанции, вспомогательных веществ, системы упаковки (укупорки) и любого релевантного дозирующего устройства), процесс производства и при необходимости сведения, полученные при разработке аналогичного лекарственного препарата.
21. Следует обосновать применение любых диапазонов содержания вспомогательных веществ, включенных в производственную рецептуру, с учетом опыта, полученного во время разработки или производства лекарственного препарата.
22. Следует представить резюме составов, использованных в клинических исследованиях безопасности и эффективности и любых соответствующих исследованиях биодоступности или биоэквивалентности. Следует подробно описать любые изменения между составом и лекарственной формой лекарственного препарата для промышленного производства и составом и лекарственной формой серий, использованных в опорных клинических исследованиях, а также составом и лекарственной формой первичных серий, произведенных для исследования стабильности, промышленными сериями и представить обоснования таких изменений.
23. Следует обобщить сведения, полученные по результатам сравнительных исследований in vitro (например, теста сравнительной кинетики растворения) или сравнительных исследований in vivo (например, исследования биоэквивалентности), которые связывают состав и лекарственную форму серий лекарственных препаратов для клинических исследований с предлагаемым составом и лекарственной формой лекарственного препарата для промышленного производства, описанными в разделе 3.2.P.1 регистрационного досье, и представить перекрестные ссылки на исследования (с указанием номеров исследований).
Если предпринимались попытки установления корреляции in vitro - in vivo, то в разделе 3.2.P.1 регистрационного досье следует указать результаты таких исследований и перекрестные ссылки на них (с указанием номеров исследований). Успешная корреляция может способствовать выбору соответствующих критериев приемлемости для теста "растворение" и может потенциально исключить необходимость проведения дальнейших исследований биоэквивалентности после любых значимых изменений лекарственного препарата (например, состава, лекарственной формы, физических характеристик лекарственной формы и др.) или процесса его производства.
24. Следует указать любые специфичные особенности дизайна лекарственного препарата (например, наличие линии разлома (риски) на таблетке, избыток массы или объема наполнения единицы дозирования лекарственного препарата, меры по борьбе с фальсификацией лекарственного препарата, если эти особенности дизайна влияют на лекарственный препарат) и представить обоснование их использования.
Производственные избытки
25. Не рекомендуется использовать производственные избытки активной фармацевтической субстанции для компенсации ее деградации во время процесса производства или в течение срока годности (срока хранения) лекарственного препарата либо для продления срока годности (срока хранения).
26. Любые избытки при производстве лекарственного препарата (попадают они в окончательный состав препарата или нет) следует обосновывать с точки зрения безопасности и эффективности лекарственного препарата. Следует представить следующие сведения:
количество производственных избытков;
причины наличия избытков (например, создание избытков, чтобы компенсировать ожидаемые и документированные производственные потери);
обоснование количества избытков.
Избытки следует включить в количественный состав лекарственного препарата, указанный в составе на серию (производственной рецептуре) (в подраздел 3.2.P.3.2 регистрационного досье).
Физико-химические и биологические свойства
27. Следует указать и рассмотреть физико-химические и биологические свойства, влияющие на безопасность, функциональные характеристики или пригодность для технологического процесса лекарственного препарата. Указанные свойства включают в себя связь показателей качества действующего вещества и лекарственной формы с их биологическим действием. Исследования могут включать в себя, например, разработку испытания на респирабельную фракцию ингаляционного лекарственного препарата. В подраздел 3.2.P.2.2.3 регистрационного досье допускается включать сведения, обосновывающие выбор между испытанием на растворение или распадаемость либо другими способами обеспечения высвобождения действующего вещества, а также разработку и пригодность выбранного испытания с учетом требований приложения N 1 к Руководству по нормативному документу (схема решений N 7 (часть 3) и схема решений N 3 (часть 1)) и положений главы 6 Правил исследований биологических лекарственных средств. В обосновании разработки лекарственного препарата следует привести ссылки на любые относящиеся к процессу разработки данные исследований стабильности, включенные в подраздел 3.2.P.8.3 регистрационного досье.
3. Разработка производственного процесса
28. Следует обосновать выбор, систему контроля и любое усовершенствование производственного процесса, описанного в подразделе 3.2.P.3.3 регистрационного досье (предназначенного для промышленных серий). Следует рассматривать критичные показатели вместе с доступными вариантами производственного процесса, чтобы объяснить выбор этого процесса и подтвердить целесообразность компонентов. Следует описать пригодность оборудования, используемого для планируемых к производству лекарственных препаратов. Исследования по разработке технологического процесса должны служить основой для совершенствования процесса производства, валидации этого процесса, непрерывной верификации процесса производства (если применимо) и любых требований в части контроля процесса производства. Такие исследования (если применимо) должны включать в себя изучение микробиологических, а также физических и химических показателей лекарственного препарата. Сведения, полученные по результатам исследований по разработке технологического процесса, можно использовать (при необходимости) для обоснования спецификации лекарственного препарата в подразделе 3.2.P.5.6 регистрационного досье.
29. В программе разработки процесса производства или программе совершенствования процесса производства следует определить любые критические параметры технологического процесса, требующие проведения мониторинга или осуществления контроля (например, конечная точка гранулирования), для обеспечения заданного качества лекарственного препарата.
30. В отношении лекарственных препаратов, которые должны быть стерильными, следует выбрать подходящий метод стерилизации лекарственного препарата и материала первичной упаковки и обосновать такой выбор.
31. Следует описать и проанализировать существенные различия между производственными процессами, которые использовались для производства серий лекарственного препарата для опорных клинических исследований (безопасности, эффективности, биодоступности, биоэквивалентности) или первичных исследований стабильности, и производственным процессом, описанным в подразделе 3.2.P.3.3 регистрационного досье. Анализ должен обобщать влияние различий между производственными процессами на функциональные характеристики, возможность производства и качество лекарственного препарата. Информацию следует представлять таким образом, чтобы облегчить сравнение процессов производства и соответствующих данных анализа серий (подраздел 3.2.P.5.4). Такая информация включают в себя, например:
а) идентификацию (например, номер серии) и назначение произведенных серий (например, номер серии для исследования биоэквивалентности);
б) наименование производственной площадки;
в) размер серии;
г) любые существенные различия в оборудовании (например, дизайн, принцип работы, размер).
32. Для обеспечения гибкости при совершенствовании технологического процесса при описании разработки процесса производства следует описать системы измерений, позволяющие вести мониторинг критических показателей или конечных точек процесса производства. Сбор данных мониторинга технологического процесса во время разработки процесса производства может позволить получить полезную информацию и лучше понять процесс. Следует описать стратегии контроля процесса производства, дающие возможность корректировать процесс производства, для обеспечения контроля всех критических показателей.
33. Допускается представить оценку способности процесса производства надежно производить лекарственный препарат с планируемым качеством (например, функционирование процесса производства при разных рабочих условиях, разных масштабах производства серии или с разным оборудованием). Понимание робастности (устойчивости) процесса производства может быть полезно для оценки и снижения риска и позволяет обосновать совершенствование процесса производства и технологического процесса (особенно в сочетании с использованием инструментов управления рисками), в соответствии с главой 2 части III Правил производственной практики.
4. Система упаковки (укупорки)
34. Следует описать и обосновать выбор системы упаковки (укупорки) для промышленной серии лекарственного препарата, описанной в разделе 3.2.P.7 регистрационного досье. Следует рассмотреть планируемое назначение лекарственного препарата и пригодность системы упаковки (укупорки) для его хранения и транспортировки (перевозки) (включая оценку упаковки нефасованного лекарственного препарата (где применимо)).
35. Следует обосновать выбор материалов первичной упаковки, в том числе описать исследования, выполненные для подтверждения целостности контейнера и укупорки. Следует рассмотреть возможное взаимодействие между лекарственным препаратом и контейнером или его маркировкой.
36. При выборе первичной упаковки следует учитывать, например, выбор материалов упаковки, защиту от влаги и света, совместимость материалов конструкции с лекарственной формой (включая сорбцию контейнером и вымывание) и безопасность материалов конструкции. В соответствующих случаях следует обосновать выбор материалов вторичной упаковки.
37. Если используется дозирующее устройство (например, пипетка-капельница, шприц-ручка, ингалятор), следует продемонстрировать воспроизводимую и точную доставку дозы лекарственного препарата в условиях испытаний, которые в максимально возможной степени имитируют применение лекарственного препарата.
5. Микробиологические характеристики
38. В подразделе 3.2.P.2.5 регистрационного досье следует описать микробиологические характеристики лекарственного препарата (если применимо). Такое описание включает в себя, например:
а) обоснование проведения или непроведения испытаний на микробиологическую чистоту для нестерильных лекарственных препаратов (например, схема решений N 2 приложения N 1 к Руководству по нормативному документу и положения главы 6 Правил исследований биологических лекарственных средств);
б) сведения о выборе и эффективности систем консервантов в лекарственных препаратах, содержащих антимикробный консервант;
в) сведения о антимикробной эффективности лекарственных препаратов, которые обладают собственным противомикробным действием;
г) обеспечение целостности системы упаковки (укупорки) стерильных лекарственных препаратов, поскольку она необходима для предотвращения микробной контаминации.
39. Несмотря на то что химическое испытание на содержание консерванта, как правило, является показателем, который включается в спецификацию лекарственного препарата, во время разработки следует подтвердить эффективность антимикробного консерванта. Используя данные, полученные в ходе испытаний на эффективность антимикробного консерванта, следует подтвердить, что наименьшая установленная концентрация антимикробного консерванта эффективна в отношении контроля содержания микроорганизмов. Используемую концентрацию следует обосновать с позиций эффективности и безопасности в целях применения минимальной концентрации консерванта, обеспечивающей требуемый уровень эффективности на протяжении всего планируемого срока годности (срока хранения) лекарственного препарата. Если целесообразно, во время разработки следует выполнить микробную провокационную пробу в условиях испытаний, которые, насколько это возможно, максимально имитируют применение лекарственного препарата пациентом, и представить ее результаты в подразделе 3.2.P.2.5 регистрационного досье.
6. Совместимость
40. Для обоснования необходимой и экспериментально подтвержденной информации о лекарственном препарате следует изучить сведения о совместимости лекарственного препарата с жидкостями для восстановления (например, осаждение, стабильность) и включить их в подраздел 3.2.P.2.6 регистрационного досье. Эти сведения включают в себя рекомендуемый срок годности (срок хранения) готового к применению лекарственного препарата при рекомендуемой температуре хранения и в вероятных крайних значениях концентрации. Также может потребоваться изучение смешивания или разведения лекарственных препаратов перед введением (например, добавление лекарственного препарата в инфузионные контейнеры большого объема).
V. Подходы к фармацевтической разработке
41. Лекарственный препарат следует проектировать таким образом, чтобы удовлетворить потребности пациентов и обеспечить соответствие лекарственного препарата его целевым функциональным характеристикам. Стратегии разработки лекарственного препарата варьируются у разных фармацевтических производителей и для разных препаратов. Подход к разработке и ее масштаб могут также варьироваться и включаются в регистрационное досье. Заявитель может выбрать эмпирический или более систематизированный подходы к разработке лекарственного препарата либо их комбинации. В приложении N 1 к настоящему Руководству приводятся характеристики возможных различий между указанными подходами. Более систематизированный подход к разработке (также определяемый как "проектирование качества") может включать в себя, например, использование предварительных знаний, результаты исследований с использованием дизайна экспериментов, использование управления рисками для качества и использование управления знаниями на протяжении жизненного цикла лекарственного препарата. Указанный систематический подход может способствовать получению желаемого качества лекарственного препарата и помогает экспертам уполномоченного органа (экспертной организации) государства-члена лучше понять стратегию фармацевтической разработки, проведенной фармацевтическим производителем. Понимание характеристик лекарственного препарата и процесса производства может совершенствоваться по мере накопления сведений, полученных в течение жизненного цикла лекарственного препарата.
42. Более детальные знания о свойствах лекарственного препарата и процесса его производства могут создать основу для выработки более гибких подходов к регулированию обращения лекарственных препаратов. Степень гибкости подходов, применяемых уполномоченными органами (экспертными организациями) государств-членов, основывается на соответствующем уровне научных сведений, представленных в регистрационном досье. Основой для научных и риск-ориентированных оценок регистрационного досье и экспертизы, проводимой уполномоченным органом (экспертной организацией) государства-члена, являются сведения, полученные и представленные уполномоченному органу (экспертной организации), а не сам объем этих собранных сведений. В регистрационном досье следует представить сведения, подтверждающие, что эти знания основаны на достоверных научных принципах.
43. Фармацевтическая разработка включает в себя по меньшей мере следующие элементы:
а) формулировка целевого профиля качества лекарственного препарата, поскольку он относится к безопасности, эффективности и качеству, исходя их таких факторов, как например, путь введения, лекарственная форма, биодоступность, дозировка и стабильность;
б) выявление потенциальных критических показателей качества (КПК) лекарственного препарата в целях изучения и контроля характеристик лекарственного препарата, которые влияют на его качество;
в) определение критических показателей качества активной фармацевтической субстанции, вспомогательных веществ и др., а также выбор вида и количества вспомогательных веществ для получения лекарственного препарата желаемого качества;
г) выбор подходящего процесса производства;
д) формулировка стратегии контроля.
44. Углубленный, основанный на проектировании качества подход к разработке лекарственного препарата дополнительно включает в себя следующие элементы:
а) систематическая оценка, понимание и совершенствование состава и процесса производства, в том числе:
выявление характеристик материалов и параметров технологического процесса, которые могут влиять на критические показатели качества лекарственного препарата (например, при помощи предварительных знаний, экспериментальных работ и оценки риска);
определение функциональных зависимостей, связывающих характеристики материалов и параметры технологического процесса с критическими показателями качества лекарственного препарата;
б) использование углубленного понимания характеристик лекарственного препарата и процесса производства совместно с управлением рисками для качества в целях создания соответствующей стратегии контроля, которая может, например, включать в себя предложение проектного поля (проектных полей) и (или) выпускающих испытаний в реальном времени.
45. Более систематический подход способствует непрерывному совершенствованию и инновациям на протяжении жизненного цикла лекарственного препарата.
VI. Элементы фармацевтической разработки
46. Описанные в настоящем разделе возможные подходы к получению систематического, углубленного понимания характеристик разрабатываемого лекарственного препарата и процесса его производства, а также приведенные примеры являются иллюстративными и не предназначены для создания новых требований к регулированию обращения лекарственных средств.
1. Целевой профиль качества лекарственного препарата
47. Целевой профиль качества лекарственного препарата является основой планирования разработки лекарственного препарата. Факторы целевого профиля качества лекарственного препарата могут включать в себя:
а) планируемое назначение лекарственного препарата в клинических условиях, путь введения (способ применения), лекарственную форму, системы доставки;
б) дозировку (дозировки);
в) систему упаковки (укупорки);
г) высвобождение или доставку активной части действующего вещества и показатели, влияющие на фармакокинетические характеристики (например, растворение, аэродинамические свойства), соответствующие разрабатываемой лекарственной форме лекарственного препарата;
д) соответствующие критерии качества лекарственного препарата (например, стерильность, чистота, стабильность, высвобождение действующего вещества) для планируемого к обращению на рынке лекарственного препарата.
2. Критические показатели качества
48. Критические показатели качества, как правило, связаны с активной фармацевтической субстанцией, вспомогательными веществами, промежуточными продуктами (внутрипроизводственными материалами) и лекарственным препаратом.
49. Критические показатели качества твердых лекарственных форм для приема внутрь - это показатели, которые влияют на чистоту, дозировку, высвобождение действующего вещества и стабильность. Критические показатели качества других систем доставки могут дополнительно включать в себя дополнительные специфические характеристики лекарственных препаратов, такие как аэродинамические свойства ингаляционных лекарственных препаратов, стерильность парентеральных лекарственных препаратов и адгезионные свойства трансдермальных пластырей. В отношении фармацевтических субстанций, исходного сырья и промежуточных продуктов критические показатели качества могут дополнительно включать в себя свойства, которые влияют на критические показатели качества лекарственного препарата (например, распределение частиц по размеру (гранулометрический состав), насыпная плотность).
50. Потенциальные критические показатели качества лекарственного препарата, полученные из целевого профиля качества лекарственного препарата и (или) предварительных сведений, используются в качестве направления разработки лекарственного препарата и процесса его производства. Перечень потенциальных критических показателей качества может модифицироваться после выбора состава и процесса производства, а также по мере накопления информации о лекарственном препарате и понимания процесса его производства. Для установления приоритетов в перечне потенциальных критических показателей качества с целью их последующей оценки можно использовать управление рисками для качества. Соответствующие критические показатели качества можно выявить при помощи итеративного процесса управления рисками для качества и экспериментальных исследований, которое оценивает, в какой степени их вариабельность может влиять на качество лекарственного препарата.
3. Оценка рисков: установление связи характеристик
материалов и параметров процесса с критическими показателями
качества лекарственного препарата
51. Оценка рисков - научный метод, используемый в области управления рисками для качества (в соответствии с частью III Правил производственной практики), который может выявить, какие характеристики материалов и параметры технологического процесса потенциально влияют на критические показатели качества лекарственного препарата. Оценка рисков, как правило, проводится на раннем этапе процесса фармацевтической разработки и повторяется по мере увеличения объема сведений и получения более подробной информации о лекарственном препарате.
52. Инструменты оценки рисков можно использовать для выявления и ранжирования параметров (например, параметров процесса, оборудования, исходных материалов), имеющих потенциал влияния на качество лекарственного препарата, на основании предварительных сведений и первоначальных экспериментальных данных. Иллюстративный пример приведен согласно приложению N 2 к настоящему Руководству. Первоначальный перечень потенциальных параметров может подвергаться модификации и приоритизации в ходе дальнейших исследований (например, посредством комбинирования дизайна экспериментов, механистических моделей). Перечень может уточняться по результатам экспериментальных работ в целях определения значимости отдельных параметров и потенциальных взаимодействий. По мере выявления значимых параметров их следует изучить более детально (например, посредством комбинирования дизайна экспериментов, математических моделей или исследований, ведущих к механистическому пониманию критических показателей качества) в целях более глубокого понимания процесса производства лекарственных препаратов.
4. Проектное поле
53. Зависимость между входными факторами процесса производства лекарственных препаратов (характеристиками материалов и параметрами процесса производства) и критическими показателями качества описывается в проектном поле (примеры применения инструментов фармацевтической разработки приведены в приложении N 2 к настоящему Руководству). Работа в пределах проектного поля не рассматривается в качестве изменения. Выход за пределы проектного поля рассматривается в качестве изменения и, как правило, влечет за собой внесение пострегистрационных изменений. Проектное поле предлагается заявителем и является объектом экспертизы уполномоченного органа (экспертной организации) государства-члена и последующего одобрения при регистрации или выполнении иных процедур, связанных с регистрацией лекарственного препарата.
54. Оценка рисков и эксперименты в ходе разработки процесса производства, описанные в подразделе 3 настоящего раздела, позволяют проанализировать связь и влияние параметров процесса производства и показателей материалов на критические показатели качества лекарственного препарата, а также выявить переменные и их диапазоны, в пределах которых может достигаться постоянство качества лекарственного препарата. Эти параметры технологического процесса и показатели материалов включаются в проектное поле.
55. В регистрационном досье следует представить описание параметров технологического процесса и характеристик материалов, рассмотренных для проектного поля, и их влияние на качество лекарственного препарата. Следует представить обоснование их включения в проектное поле. В некоторых случаях также следует представить обоснование исключения некоторых параметров. Сведения, полученные в ходе исследований, следует включать в регистрационное досье. Следует указать параметры технологического процесса и характеристики материалов, которые не варьировали на протяжении разработки.
Описание проектного поля в регистрационном досье
56. Проектное поле следует описать с точки зрения диапазонов характеристик материалов и параметров технологического процесса или при помощи более сложных математических зависимостей. Проектное поле описывается как зависящая от времени функция (например, температурный цикл и цикл давления в цикле лиофилизации) или комбинация переменных, например, компонентов многомерной (многофакторной, многовариантной) модели. Допускается также включить в регистрационное досье факторы масштабирования, если проектное поле предназначено охватить несколько операционных масштабов. Анализ ретроспективных данных следует учитывать при установлении проектного поля. Независимо от способов разработки проектного поля, ожидается, что работа в пределах проектного поля будет приводить к получению лекарственного препарата, соответствующего заданному качеству.
57. Примеры разных потенциальных подходов к представлению проектного поля приведены в разделе III приложения N 2 к настоящему Руководству.
Проектное поле (проектные поля) единичной операции
58. Заявитель вправе установить независимые проектные поля для одной или более единичных операций либо единое проектное поле, охватывающее несколько операций. Несмотря на то, что отдельное проектное поле для каждой единичной операции разработать как правило проще, разработка проектного поля, охватывающего процесс производства лекарственного препарата целиком, может давать  операционную гибкость. Например, для лекарственного препарата, подвергающегося деградации в растворе перед лиофилизацией, проектное поле для контроля степени деградации (например, концентрация, время, температура) может быть выражено для каждой единичной операции или суммы всех единичных операций.
операционную гибкость. Например, для лекарственного препарата, подвергающегося деградации в растворе перед лиофилизацией, проектное поле для контроля степени деградации (например, концентрация, время, температура) может быть выражено для каждой единичной операции или суммы всех единичных операций.
Зависимость проектного поля от масштаба производства
и оборудования
59. При описании проектного поля заявителю следует учесть вид желаемой операционной гибкости. Проектное поле может быть разработано при любом масштабе. Заявителю следует:
обосновать возможность применения для производственного процесса с предлагаемым масштабом производства проектного поля, разработанного для малого и опытно-промышленного масштаба производства;
описать и проанализировать потенциальные риски операции масштабирования.
60. Если заявитель предлагает единое проектное поле, применимое к нескольким операционным масштабам, такое поле необходимо описать с точки зрения релевантных, не зависящих от масштаба параметров. Например, если установлено, что лекарственный препарат чувствителен к сдвигу во время операции смешивания, проектное поле может включать в себя скорость сдвига, а не скорость перемешивания. В описание проектного поля можно включить безразмерные цифры и (или) модели для масштабирования.
Проектное поле и доказанные приемлемые диапазоны
61. Комбинация доказанных приемлемых диапазонов не образует проектного поля. Доказанные приемлемые диапазоны, основанные на одномерном экспериментировании, могут служить источником полезных сведений о процессе производства.
Проектное поле и граница отказа
62. Следует определить границу отказа для параметров технологического процесса или характеристик материалов, перейдя которую невозможно обеспечить достижение соответствующих показателей качества. Вместе с тем определение границы отказа или установление режимов отказа не являются обязательными компонентами установления проектного поля.
5. Стратегия контроля
63. Стратегия контроля формируется для обеспечения стабильного производства лекарственного препарата заданного качества. Средства контроля могут включать в себя параметры и показатели, относящиеся к материалам и компонентам лекарственного препарата, условия эксплуатации объекта и оборудования, виды внутрипроизводственного контроля, спецификации на готовые лекарственные препараты, а также связанные методы и частоту мониторинга и контроля. Элементы стратегии контроля, включенные в раздел 3.2.P.2 регистрационного досье, должны описывать и обосновывать, как внутрипроизводственные виды контроля и средства контроля исходного сырья и материалов (активной фармацевтической субстанции и вспомогательных веществ), промежуточных продуктов (внутрипроизводственных материалов), системы упаковки (укупорки) лекарственных препаратов влияют на качество выпускаемого лекарственного препарата. Указанные виды контроля основываются на знании характеристик лекарственного препарата, его состава и процесса производства и, по меньшей мере, включают в себя контроль критических параметров процесса производства лекарственного препарата и характеристик исходного сырья и материалов.
64. Всесторонний подход к фармацевтической разработке будет обеспечивать понимание процесса производства и характеристик лекарственного препарата и выявлять источники вариабельности. Источники вариабельности, которые могут влиять на качество лекарственного препарата, следует определить, правильно интерпретировать и впоследствии контролировать. Понимание источников вариабельности и их влияния на последующие процессы производства или обработку, внутрипроизводственные материалы и качество лекарственного препарата может обеспечить возможность для переноса видов контроля на предыдущие стадии и минимизации необходимости в испытаниях при выпуске лекарственного препарата. Понимание источников вариабельности и их влияния на лекарственный препарат и процесс его производства вместе с управлением рисками для качества (в соответствии с частью III Правил производственной практики) будет обосновывать контроль процесса производства таким образом, что вариабельность (например, сырья) может быть компенсирована адаптацией режима для получения лекарственного препарата с постоянным качеством.
65. Такое понимание процесса производства может дать возможность для альтернативной производственной парадигмы, когда вариабельность исходного сырья и материалов может быть ограничена менее жестко. Вместо этого может появиться возможность спланировать адаптивную стадию процесса производства (стадию, реагирующую на исходные материалы) с соответствующим контролем процесса производства, чтобы обеспечить постоянное качество лекарственного препарата.
66. Углубленное понимание функциональных характеристик лекарственного препарата может обосновывать использование альтернативных подходов для определения соответствия исходного сырья и материалов показателям качества. Использование подобных альтернатив может обосновывать испытания в реальном времени. Например, тест "Распадаемость" может служить косвенным подтверждением растворения быстрораспадающихся твердых лекарственных форм с хорошо растворимыми активными фармацевтическими субстанциями. Внутрипроизводственное испытание на однородность дозированных единиц (например, использование однородности массы совместно с анализом в ближнем инфракрасном диапазоне спектра (БИК)) может дать возможность для проведения испытаний в реальном времени и позволить добиться повышенного уровня обеспечения качества по сравнению с проведением традиционного испытания при выпуске лекарственного препарата с использованием фармакопейных подходов. Испытания в реальном времени могут заменить испытания конечного продукта, но не заменяют стадии проверки и контроля качества, предусмотренные Правилами производственной практики в отношении выпуска серии.
67. Стратегия контроля может включать в себя в том числе следующие сведения:
а) параметры контроля характеристик исходных материалов (например, активной фармацевтической субстанции, вспомогательных веществ, материалов первичной упаковки), основанный на понимании их влияния на процесс производства или качество лекарственного препарата;
б) параметры спецификация (спецификации) лекарственного препарата;
в) параметры контроля единичных операций, влияющих на последующую обработку или качество лекарственного препарата (например, влияние сушки на деградацию, распределения частиц по размеру гранулята на растворение);
г) сведения о внутрипроизводственных испытаниях или испытаний в реальном времени вместо испытаний конечного продукта (например, об измерении и контроле критических показателей качества во время обработки);
д) программа мониторинга (например, регулярный полный анализ лекарственного препарата) в целях верификации предсказательных многофакторных моделей.
68. Стратегия контроля может включать в себя разные элементы (например, один элемент стратегии контроля может основываться на результатах испытаний конечного продукта, тогда как другой может зависеть от результатов испытаний в реальном времени). В регистрационном досье необходимо описать основание использования таких альтернативных подходов.
69. Применение положений настоящего Руководства следует использовать для обоснования альтернативных подходов к установлению показателей и критериев приемлемости спецификаций, указанных в приложении N 1 к Руководству по нормативному документу и главе 6 Правил исследований биологических лекарственных средств.
6. Управление жизненным циклом лекарственного препарата
и непрерывное совершенствование
70. На протяжении жизненного цикла лекарственного препарата фармацевтические производители имеют возможность оценить инновационные подходы к улучшению качества лекарственного препарата в соответствии с частью III Правил производственной практики.
71. Мониторинг процесса производства лекарственного препарата может проводиться в целях подтверждения того, что процесс производства осуществляется в ожидаемом режиме, необходимом для получения показателей качества лекарственного препарата, запланированных в проектном поле. Такой мониторинг может включать в себя анализ тенденций процесса производства по мере накопления дополнительного опыта во время рутинного производства. В случае определенных проектных полей с использованием математических моделей периодическое поддержание может оказаться полезным для обеспечения работы модели. Поддержание модели - пример активности, которой фармацевтический производитель может управлять в рамках собственной системы качества при условии неизменности проектного поля.
72. После получения дополнительных знаний о процессе производства может появиться необходимость расширить, сократить или переформулировать проектное поле. Изменение проектного поля следует проводить в соответствии с требованиями актов органов Союза в сфере обращения лекарственных средств.
VII. Представление фармацевтической разработки и связанных
сведений в регистрационном досье в формате общего
технического документа (ОТД)
73. Сведения о фармацевтической разработке включаются в раздел 3.2.P.2 регистрационного досье. Иные сведения, получаемые по результатам исследований по фармацевтической разработке, допускается распределить по регистрационному досье несколькими способами. Вместе с тем заявителю следует четко указать, где располагаются разные сведения. Помимо того, что включено в регистрационное досье, определенные аспекты (например, управление жизненным циклом лекарственного препарата, непрерывное совершенствование) настоящего Руководства рассматриваются в рамках фармацевтической системы качества заявителя в соответствии с частью III Правил производственной практики.
1. Управление рисками для качества и разработка
лекарственного препарата и процесса его производства
74. Управление рисками для качества следует использовать на разных стадиях разработки лекарственного препарата и процесса производства. Результаты проведенной оценки, использованные для ориентирования и обоснования решений по разработке, можно включить в соответствующие подразделы раздела 3.1.P.2 регистрационного досье. Например, анализы рисков и функциональные зависимости, позволяющие связать показатели материалов и параметры процесса производства с критическими показателями качества лекарственного препарата, можно включить в подразделы 3.2.P.2.1, 3.2.P.2.2 и 3.2.P.2.3 регистрационного досье. Анализы рисков, позволяющие связать дизайн процесса производства с качеством лекарственного препарата, можно включить в подраздел 3.2.P.2.3 регистрационного досье.
2. Проектное поле
75. В качестве элемента предлагаемого процесса производства проектное поле (проектные поля) можно описать в подразделе 3.2.P.3.3 регистрационного досье, включающем в себя описание производственного процесса и его контроля. Дополнительные сведения (если обосновано) могут использоваться в подразделе 3.2.P.3.4 регистрационного досье, в котором рассматриваются средства контроля критических стадий и промежуточной продукции. Подразделы 3.2.P.2.1, 3.2.P.2.2 и 3.2.P.2.3 регистрационного досье подходят для обобщения и описания исследований по разработке лекарственного препарата и производственного процесса, служащих основой для проектного поля (полей). Зависимость проектного поля (проектных полей) от общей стратегии контроля следует описать в подразделе 3.2.P.5.6 регистрационного досье, включающем в себя обоснование спецификаций на лекарственный препарат.
3. Стратегия контроля
76. Общая стратегия контроля лекарственного препарата резюмируется в подразделе 3.2.P.5.6 регистрационного досье, в который включено обоснование спецификаций на лекарственный препарат. Вместе с тем подробные сведения о контроле исходного сырья и материалов и средствах контроля процесса производства следует представлять в соответствующих разделах регистрационного досье (например, для активной фармацевтической субстанции (раздел 3.2.S), контроля качества вспомогательных веществ (раздел 3.2.P.4), описания производственного процесса и его контроля (подраздел 3.2.P.3.3), контроля критических стадий и промежуточной продукции (подраздел 3.2.P.3.4)).
4. Сведения, относящиеся к активной
фармацевтической субстанции
77. Если критические показатели качества активной фармацевтической субстанции обладают потенциалом влиять на критические показатели качества или процесс производства лекарственного препарата, в подразделе 3.2.P.2.1 регистрационного досье по фармацевтической разработке целесообразно представлять анализ характеристик качества активной фармацевтической субстанции.
Приложение N 1
к Руководству по фармацевтической
разработке лекарственных средств
ХАРАКТЕРИСТИКА
РАЗЛИЧИЙ МЕЖДУ МИНИМАЛЬНЫМ И УГЛУБЛЕННЫМ ПОДХОДОМ
К ФАРМАЦЕВТИЧЕСКОЙ РАЗРАБОТКЕ
Сведения, представленные в таблице, составлены для иллюстрации некоторых потенциальных различий между минимальным подходом и углубленным, основанным на проектировании качества в отношении разных аспектов фармацевтической разработки и управления жизненным циклом лекарственного препарата. Сравнения представлены с целью улучшения понимания спектра потенциальных подходов к фармацевтической разработке, поэтому их не следует рассматривать в качестве исчерпывающих. Таблица не предназначена для того, чтобы целенаправленно сформулировать единственно возможный подход, который может выбрать производитель лекарственного препарата. При применении углубленного подхода не является обязательным установление проектного поля или проведение испытания при выпуске в реальном времени. Текущие практики, применяемые в фармацевтической отрасли, отличаются у разных производителей и обычно представляют собой среднее двух подходов, представленных в таблице.
Таблица
Аспект
Минимальные подходы
Углубленные подходы, основанные на проектировании качества
Фармацевтическая разработка в целом
-
преимущественно эмпирическая
-
систематический подход, соотносящий механистическое понимание показателей исходного сырья и материалов и параметров процесса производства с критическими показателями качества лекарственного препарата
-
исследования по разработке часто проводятся по одной переменной за раз
-
многофакторные эксперименты для понимания назначения лекарственного препарата и процесса производства
-
установление проектного поля
-
использование инструментов процессно-аналитической технологии
Процесс производства
- фиксированный
-
корректируемый в пределах проектного поля
-
валидация преимущественно основана на первоначальных полномасштабных сериях
-
подход к валидации на основании жизненного цикла и (в идеальном случае) непрерывная верификация процесса производства
-
нацеленность на оптимизацию и воспроизводимость
-
нацеленность на стратегию контроля и устойчивость
-
использование статистических методов контроля процесса
Контроль процесса производства
-
внутрипроизводственные испытания преимущественно для принятия решений "пропустить (не пропустить)" (годен (негоден))
-
использование инструментов процессно-аналитической технологии с соответствующими прямым и обратным видами контроля
-
операции процесса производства отслеживаются и формируются тенденции для содействия непрерывного совершенствования контроля процесса производства после регистрации
-
анализ вне производственной линии
Спецификации лекарственного препарата
-
основные способы контроля
-
часть общей стратегии контроля качества
-
спецификации основаны на данных о сериях, доступных на момент регистрации
-
спецификации основаны на желаемых функциональных характеристиках лекарственного препарата с релевантными обосновывающими данными
Стратегия контроля
-
качество лекарственного препарата контролируется преимущественно путем испытаний промежуточных продуктов (внутрипроизводственных материалов) и конечного продукта
-
качество лекарственного препарата обеспечивается основанной на управлении рисками стратегией контроля хорошо понимаемых характеристик лекарственного препарата и процесса производства
-
средства контроля качества переносятся на ранние стадии с возможностью проведения испытаний при выпуске в реальном времени или сокращенных испытаний конечного продукта
Управление жизненным циклом
-
реактивный (решение проблем по мере их возникновения и корректирующие действия)
-
превентивные действия
-
содействие непрерывному совершенствованию
Приложение N 2
к Руководству по фармацевтической
разработке лекарственных средств
ПРИМЕРЫ
ПРИМЕНЕНИЯ ИНСТРУМЕНТОВ ФАРМАЦЕВТИЧЕСКОЙ РАЗРАБОТКИ
I. Инструмент оценки рисков
Междисциплинарная команда экспертов может совместно разработать диаграмму Исикавы (диаграмма "рыбий скелет") (рисунок 1) для выявления потенциальных переменных, способных влиять на желаемую характеристику качества. После этого команда может ранжировать переменные на основании вероятности, тяжести и выявляемости с использованием анализа режимов и последствий отказов (FMEA) или аналогичных инструментов, основываясь на предварительных знаниях и первоначальных экспериментальных данных. Затем можно использовать дизайн экспериментов или иные экспериментальные подходы, чтобы оценить влияние переменных с высоким рангом, достичь лучшего понимания технологического процесса и разработать надлежащую стратегию контроля.
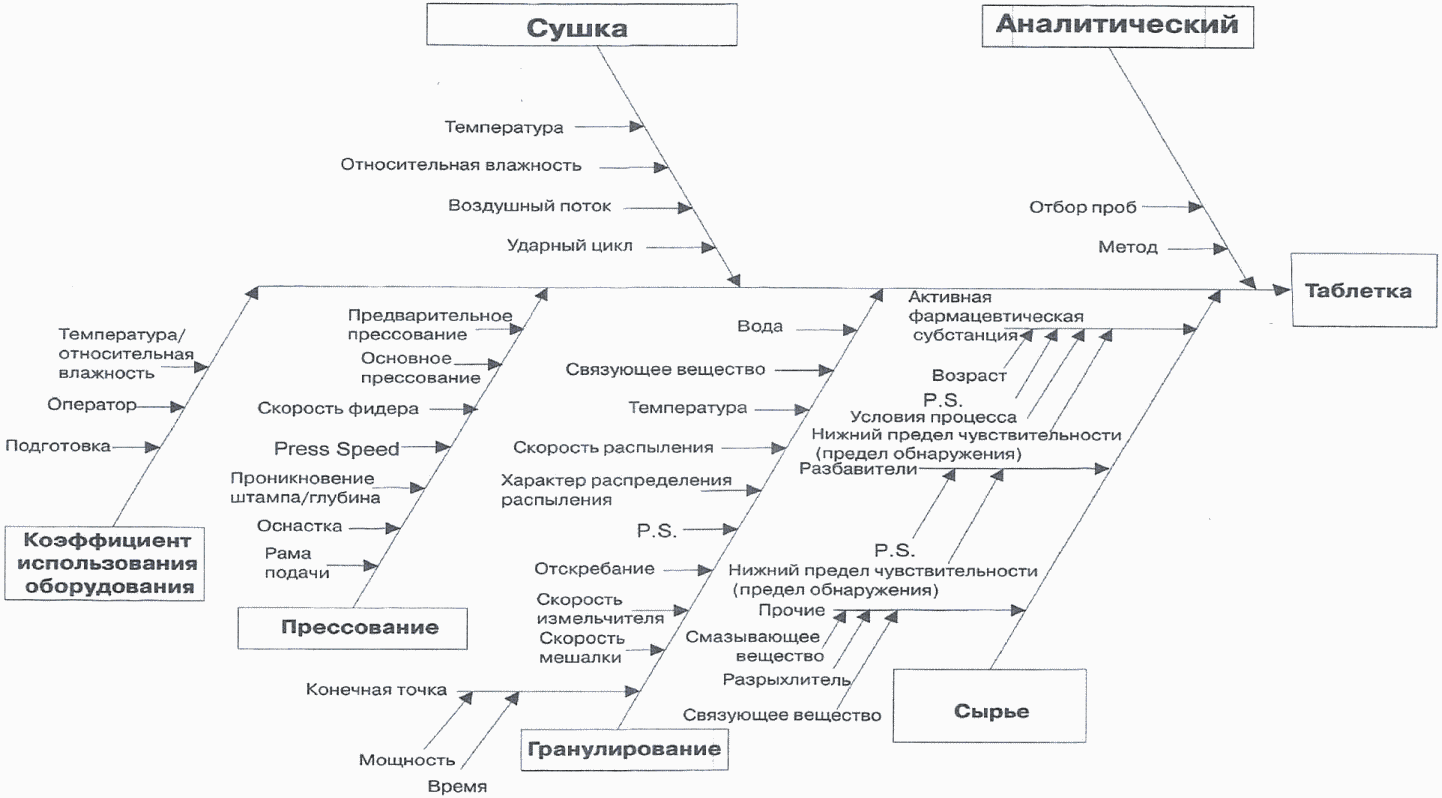
Рисунок 1. Диаграмма Исикавы
II. Отражение взаимодействий
Рисунок 2 иллюстрирует наличие или отсутствие влияния взаимодействий между тремя параметрами технологического процесса на содержание продукта деградации Y. Рисунок показывает серию двумерных графиков, отражающих влияние взаимодействий между тремя параметрами технологического процесса (исходным содержанием влаги, температурой, средним размером частиц) сушки гранулята (промежуточный продукт лекарственного препарата) на продукт деградации Y. Относительные наклоны линий или кривых на графике указывают на наличие взаимодействия. В данном примере взаимодействуют исходное содержание влаги и температура, а исходное содержание влаги и средний размер частиц - нет, равно как и температура и средний размер частиц.
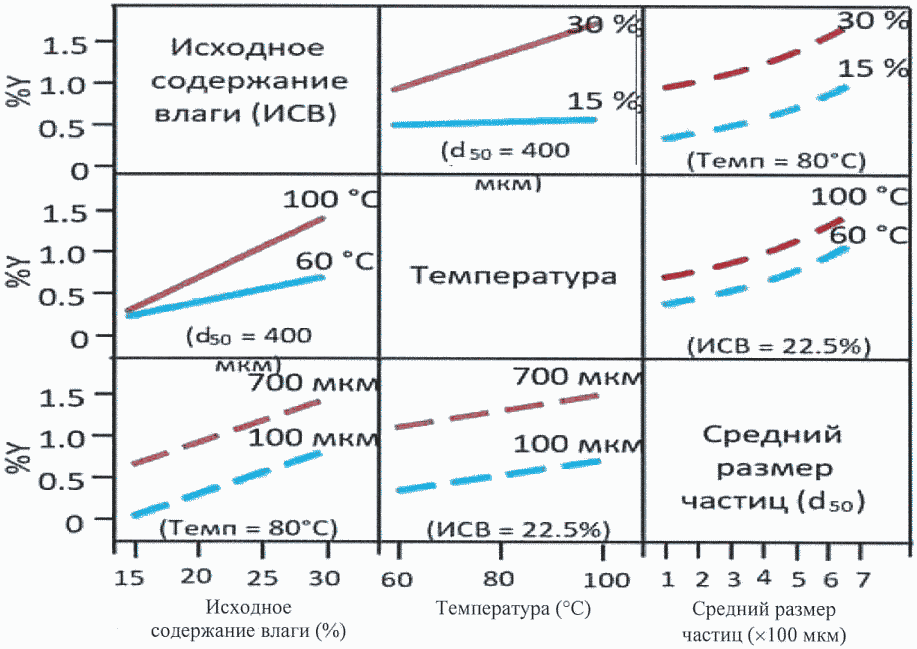
Рисунок 2. Графики влияния изменения технологических
параметров на содержание примеси в составе лекарственного
препарата
III. Представление проектного поля
Пример 1. Графики степени высвобождения действующего вещества изображены в виде диаграммы поверхности (рисунок 3) и контурной диаграммы (рисунок 4). Параметры 1 и 2 являются факторами операции гранулирования, которые влияют на степень высвобождения действующего вещества из таблетки (например, характеристика вспомогательного вещества, содержание воды, размер гранул).
На рисунках 5 и 6 приводятся два примера потенциальных проектных полей. Проектное поле на рисунке 5 задано нелинейной комбинацией диапазонов параметров, оказывающей влияние на степень высвобождения действующего вещества. В этом примере проектное поле выражается с помощью уравнения поверхности степени высвобождения, решаемого в параметрах приемлемого уровня высвобождения (80%). Приемлемый диапазон одного параметра зависит от значения другого, например:
если значение параметра 1 равно 46, то диапазон параметра 2 задается между 0 и 1,5;
если значение параметра 2 равно 0,8, то диапазон параметра 1 задается между 43 и 54.
Подход, представленный на рисунке 5, позволяет добиться максимального рабочего диапазона для достижения желаемой степени высвобождения. На рисунке 6 проектное поле задается меньшим диапазоном, основанным на линейной комбинации параметров:
диапазон параметра 1 задан между 44 и 53;
диапазон параметра 2 задан между 0 и 1,1.
Поскольку подход, представленный на рисунке 6, является более ограничивающим, заявитель вправе выбрать его вследствие простоты операции.
В этом примере рассматриваются только два параметра, поэтому их несложно представить графически. При наличии нескольких параметров проектное поле для двух параметров можно представить способом, аналогичным представленным выше примерам, в случае получения других значений (например, высоких, средних, низких) - в пределах диапазона третьего параметра, четвертого параметра и др. Альтернативно проектное поле можно выразить математически с помощью уравнений, описывающих зависимости между параметрами для успешной операции.
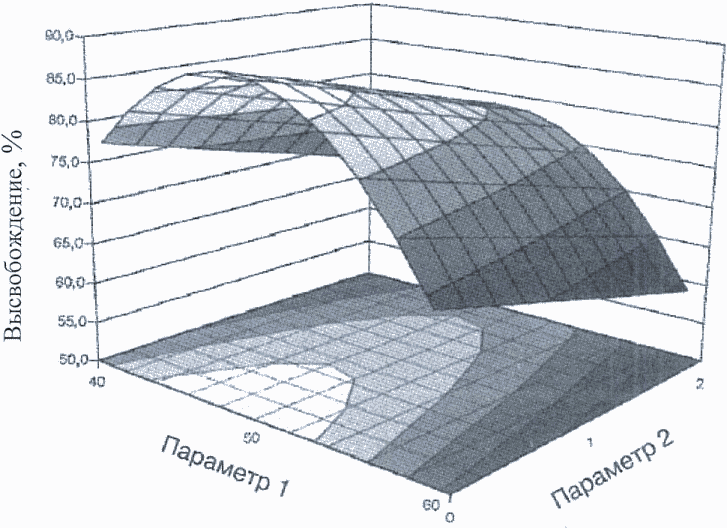
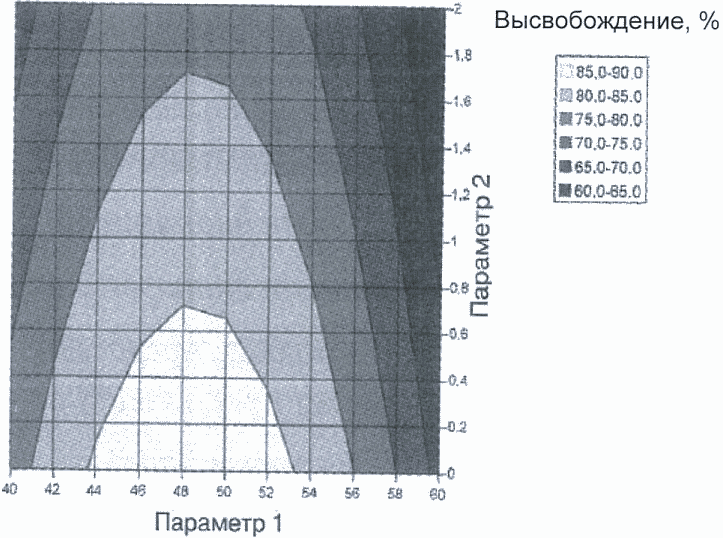
Рисунок 3. Диаграмма поверхности степени высвобождения действующего вещества как функция двух параметров операции гранулирования. Приемлемая степень высвобождения - более 80%
Рисунок 4. Контурная диаграмма степени высвобождения из примера
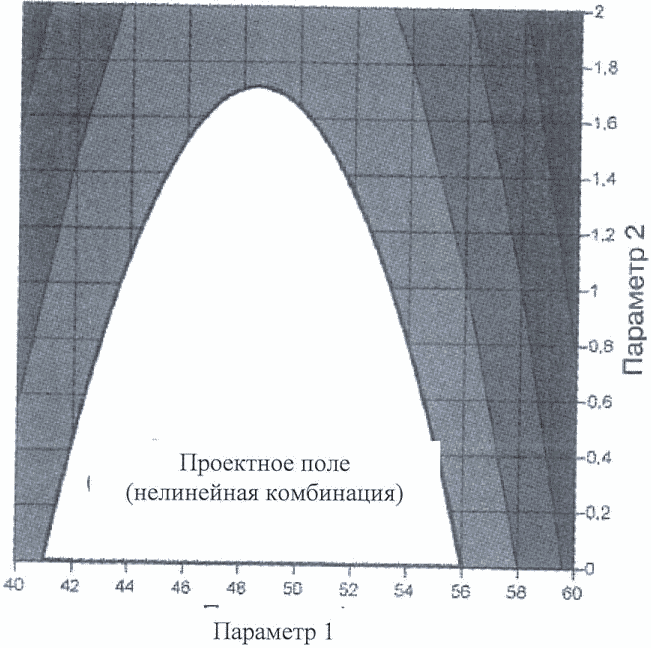
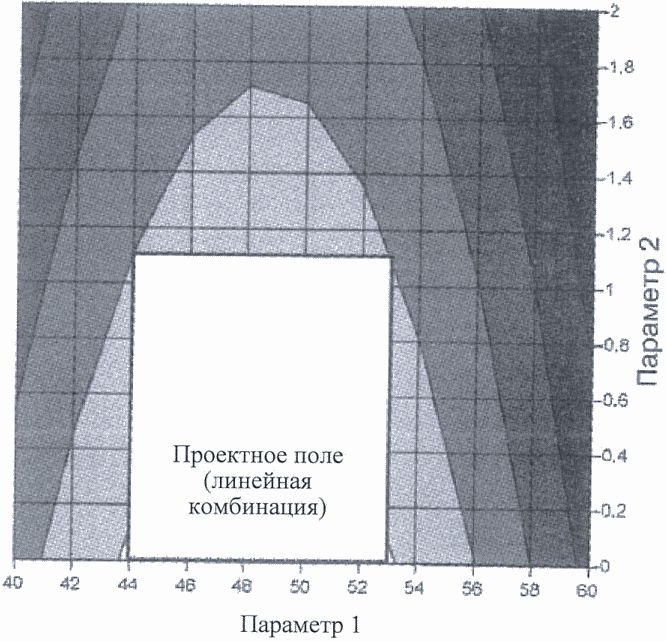
Рисунок 5. Проектное поле для определения параметров гранулирования, заданное нелинейной комбинацией их диапазонов, обеспечивающей приемлемую степень высвобождения (более 80%)
Рисунок 6. Проектное поле для определения параметров гранулирования, заданное линейной комбинацией их диапазонов, обеспечивающей приемлемую степень высвобождения (более 80%).
Пример 2. Проектное поле определено на общем участке успешных операционных диапазонов для нескольких критических показателей качества. Зависимость двух критических показателей качества (истираемость таблеток и степень высвобождения действующего вещества) от двух параметров процесса гранулирования показаны на рисунках 7 и 8. Параметры 1 и 2 представляют собой факторы операции гранулирования, влияющие на степень высвобождения действующего вещества из таблетки (например, характеристика вспомогательного вещества, содержание воды, размер гранул). Рисунок 9 показывает наложение этих участков и максимальные диапазоны предлагаемого проектного поля. В качестве проектного поля заявитель может выбрать всю область или ее часть.
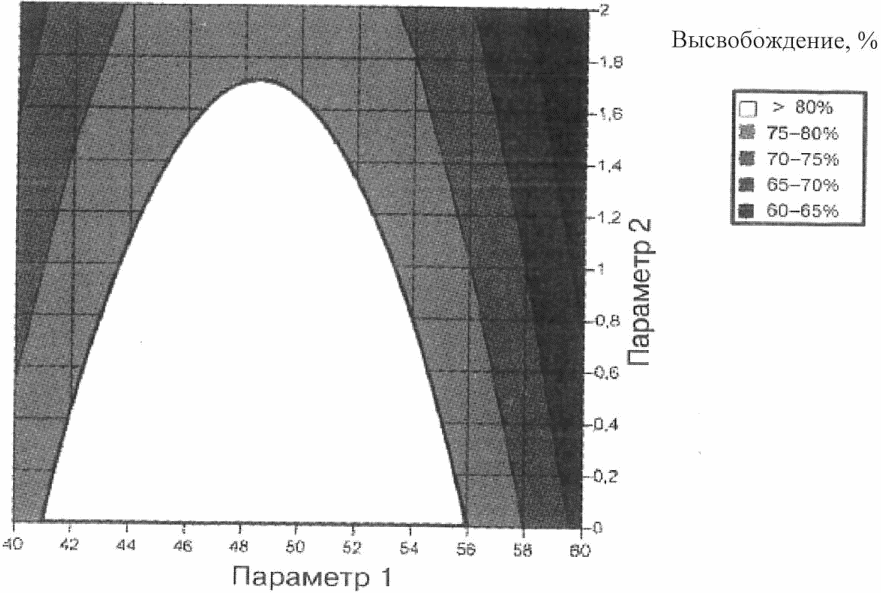
Рисунок 7. Контурная диаграмма высвобождения как функция
параметров 1 и 2
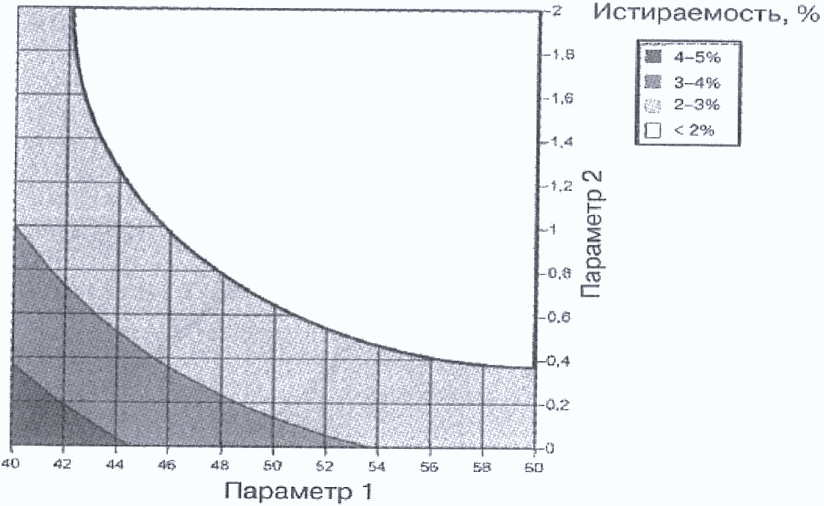
Рисунок 8. Контурная диаграмма истираемости как функция
параметров 1 и 2
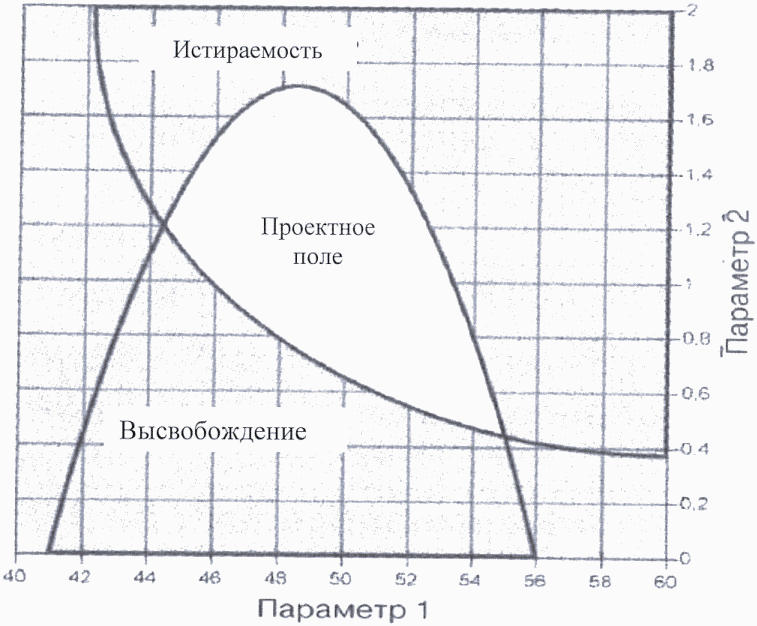
Рисунок 9. Предлагаемое проектное поле
(представляет собой область наложения диапазонов
истираемости и степени высвобождения)
Пример 3. Проектное поле для операции сушки, которая зависит от динамики температуры и (или) давления во времени. Конечная точка для содержания остаточной влаги составляет 1 - 2%. Работа выше верхнего предела проектного поля может привести к избыточному образованию примесей, а работа ниже нижнего предела проектного поля может привести к избыточному истиранию частиц.
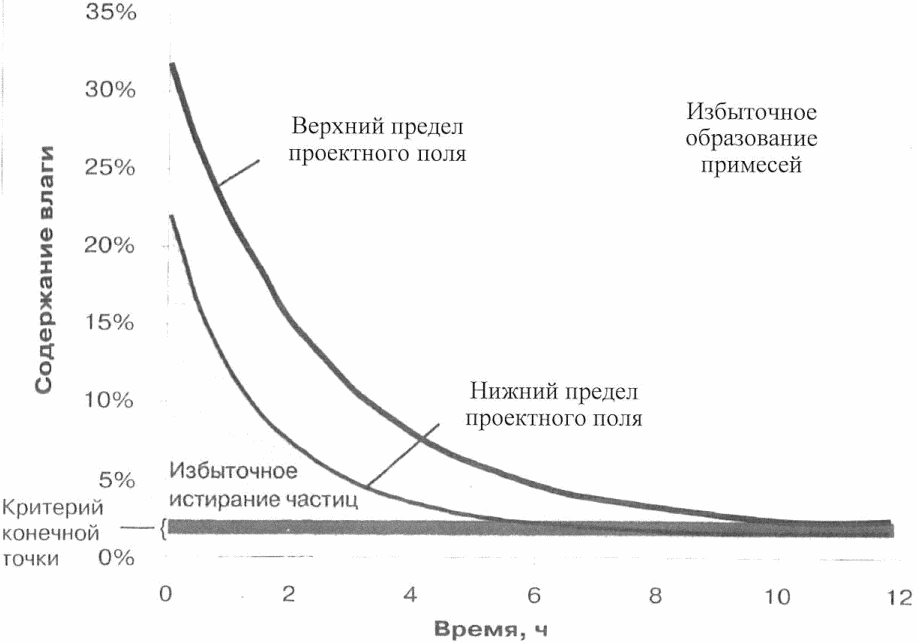
Рисунок 10. Проектное поле влияния изменений температуры
и (или) давления с течением времени на содержание
остаточной влаги