"Клинические рекомендации "Недостаточность митохондриальной ацетоацетил-КоА-тиолазы (дефицит бета-кетотиолазы, дефицит Т2)"

МИНИСТЕРСТВО ЗДРАВООХРАНЕНИЯ РОССИЙСКОЙ ФЕДЕРАЦИИ
КЛИНИЧЕСКИЕ РЕКОМЕНДАЦИИ
НЕДОСТАТОЧНОСТЬ
МИТОХОНДРИАЛЬНОЙ АЦЕТОАЦЕТИЛ-КОА-ТИОЛАЗЫ (ДЕФИЦИТ
БЕТА-КЕТОТИОЛАЗЫ, ДЕФИЦИТ Т2)
Кодирование по Международной статистической классификации болезней и проблем, связанных со здоровьем: E71.1
Год утверждения (частота пересмотра): 2024
Возрастная категория: Взрослые, Дети
Пересмотр не позднее: 2026
ID: 789
Разработчик клинической рекомендации
- Ассоциация медицинских генетиков
- Союз педиатров России
Одобрено Научно-практическим Советом Минздрава РФ
Список сокращений
2МАА-КоА - 2-метилацетоацетил-КоА;
2М3ГБ-КоА - 2-метил-3-гидроксибутирил-КоА;
3ГБ - 3-гидроксибутират;
3ГБ-дегидрогеназа - 3-гидроксибутиратдегидрогеназа;
AcAc - ацетоацетат;
C5:1 - 3-метилкротонилкарнитин;
C5OH - 3-гидроксиизовалерилкарнитин;
HbA1c - гликированный гемоглобин;
SCOT (succinyl-CoA:3-ketoacid CoA transferase) - сукцинил-КоА 3-кетоацил-КоА-трансфераза (сукцинил КоА:3-оксокислотная КоА-трансфераза);
АА-КоА - ацетоацетил-кофермента А;
ГМГ-КоА - 3-гидрокси-3-метил-глутарил-КоА;
МАТ - митохондриальная ацетоацетил-кофермент А тиолаза (Т2);
мГМГ-КоА-синтаза - митохондриальная ГМГ-КоА-синтаза;
НМАТ - недостаточность митохондриальной ацетоацетил-кофермент А тиолазы (Т2);
Т1 - митохондриальная 3-кетоацил-КоА-тиолаза;
ТМС - тандемная масс-спектрометрия.
Термины и определения
Метаболический криз - критическое, угрожающее жизни состояние, спровоцированное неблагоприятными факторами, обуславливающими усиление процессов клеточного катаболизма с накоплением токсичных производных и проявляющееся остро возникшей энцефалопатией, приступами рвоты, судорогами.
Неонатальный скрининг - проведение массового обследования новорожденных детей на наиболее распространенные врожденные и наследственные заболевания в целях предотвращения развития тяжелых форм заболеваний до развития клинических симптомов и своевременного лечения.
1. Краткая информация по заболеванию или состоянию (группы заболеваний или состояний)
1.1 Определение заболевания или состояния (группы заболеваний или состояний)
Недостаточность митохондриальной ацетоацетил-КоА-тиолазы (недостаточность МАТ, недостаточность бета-кетотиолазы, дефицит Т2, OMIM 203750) - прогрессирующее наследственное заболевание обмена веществ, в основе которого лежит дефект гена ACAT1, связанного с нарушением метаболизма изолейцина и кетоновых тел. Заболевание характеризуется эпизодами кетоацидоза с накоплением метаболитов изолейцина, обнаруживаемых при количественном и качественном анализе органических кислот мочи (2-метил-3-гидроксибутират (2М3ГБ), 2-метилацетоацетат, тиглилглицин), а также повышением концентрации ацилкарнитинов в крови (3-гидроксиизовалерилкарнитин (C5OH) и 3-метилкротонилкарнитин (C5:1)) с гипогликемией или без нее [1].
1.2 Этиология и патогенез заболевания или состояния (группы заболеваний или состояний)
Недостаточность МАТ (НМАТ) - это генетическое заболевание, характеризующееся нарушением катаболизма изолейцина и утилизации кетоновых тел, что предрасполагает к возникновению эпизодов кетоацидоза [2]. Заболевание было впервые описано в 1971 году у 6-летнего мальчика с эпизодами метаболического ацидоза и повышенным содержанием ![]() -метилацетоацетата и
-метилацетоацетата и ![]() -метил-
-метил-![]() -гидроксибутирата в моче [3]. Характерные лабораторные данные включают выраженную кетонурию и повышенную экскрецию с мочой промежуточных продуктов катаболизма изолейцина, таких как 2-метил-3-гидроксибутират (2М3ГБ), тиглилглицин и 2-метилацетоацетат [2]. Известны случаи НМАТ с острым кетоацидозом и "метаболическим инсультом" [1]. Выявление патогенных вариантов гена ACAT1 по результатам ДНК-диагностики позволяет подтвердить диагноз НМАТ на молекулярно-генетическом уровне [4].
-гидроксибутирата в моче [3]. Характерные лабораторные данные включают выраженную кетонурию и повышенную экскрецию с мочой промежуточных продуктов катаболизма изолейцина, таких как 2-метил-3-гидроксибутират (2М3ГБ), тиглилглицин и 2-метилацетоацетат [2]. Известны случаи НМАТ с острым кетоацидозом и "метаболическим инсультом" [1]. Выявление патогенных вариантов гена ACAT1 по результатам ДНК-диагностики позволяет подтвердить диагноз НМАТ на молекулярно-генетическом уровне [4].
Метаболизм кетоновых тел начинается в гепатоцитах с бета-окисления жирных кислот и образованием ацетил-КоА и ацетоацетил-кофермента А (АА-КоА). МАТ - фермент митохондриальной матрицы - преобразовывает ацетил-КоА в АА-КоА и наоборот. Митохондриальная ГМГ-КоА-синтаза (мГМГ-КоА-синтаза) катализирует реакцию образования 3-гидрокси-3-метил-глутарил-КоА (ГМГ-КоА) из ацетил-КоА и АА-КоА. ГМГ-КоА под воздействием ГМГ-КоА-лиазы превращается в ацетоацетат (AcAc), который частично восстанавливается до 3-гидроксибутирата (3ГБ) за счет активности 3-гидроксибутиратдегидрогеназы (3ГБ-дегидрогеназы) (рис. 1). Кетоновые тела AcAc и 3ГБ в отсутствие глюкозы являются важными источниками энергии для внепеченочных органов, в частности для мозга в периоды недостаточного энергоснабжения. Далее AcAc и 3ГБ переносятся во внепеченочные ткани через кровоток, где 3ГБ превращается обратно в AcAc. Сукцинил-КоА 3-кетоацил-КоА-трансфераза (SCOT) превращает AcAc в AcAc-КоА, который затем снова расщепляется ферментом МАТ и образуется ацетил-КоА (рис. 1).
В катаболизме изолейцина фермент МАТ отвечает за превращение 2-метилацетоацетил-КоА (2МАА-КоА) в ацетил-КоА и пропионил-КоА. Реакции превращение тиглил-КоА в 2-метил-3-гидроксибутирил-КоА (2М3ГБ-КоА) и превращение 2М3ГБ-КоА в 2МАА-КоА являются обратимыми. Последняя реакция катализируется 2-метил-3-гидроксибутирил-КоА-дегидрогеназой. При НМАТ происходит накопление 2МАА-КоА, 2М3ГБ-КоА и тиглил-КоА (рис. 1).
Митохондриальная 3-кетоацил-КоА-тиолаза (Т1) компенсирует НМАТ в кетогенезе, поэтому НМАТ приводит в основном к кетозу.
Заболевание возникает в результате мутаций в гене ACAT1, кодирующем фермент МАТ. Ген картирован на длинном плече 11 хромосомы (11q22.3-23.1) и состоит из 12 экзонов, охватывающих приблизительно 27 кб. На сегодняшний день известно по меньшей мере о 105 мутациях в гене ACAT1, большинство которых являются миссенс-мутациями [5]. Так, к примеру, в китайской популяции преобладают патогенные варианты c.622C> T (p. R208*), c.1006-1G>C и c.1124A> G (p. N375S) [6], а в индийской - вариант c.578T> G (p.Met193Arg) [7]. Во Вьетнаме превалирует патогенный вариант c.622C> T (p.Arg208*) [8, 9].
В настоящее время гено-фенотипическая корреляция НМАТ не установлена [10].
1.3 Эпидемиология заболевания или состояния (группы заболеваний или состояний)
Частота НМАТ в мире составляет приблизительно 1 на 1 миллион новорожденных [11]. Во Вьетнаме частота заболевания в период с 2005 по 2016 гг. составила 1:190,000 новорожденных [8], а в Китае по результатам неонатального скрининга 16 миллионов новорожденных частота НМАТ составила 1 на 1 миллион [6]. Частота встречаемости НМАТ в Российской Федерации не установлена.
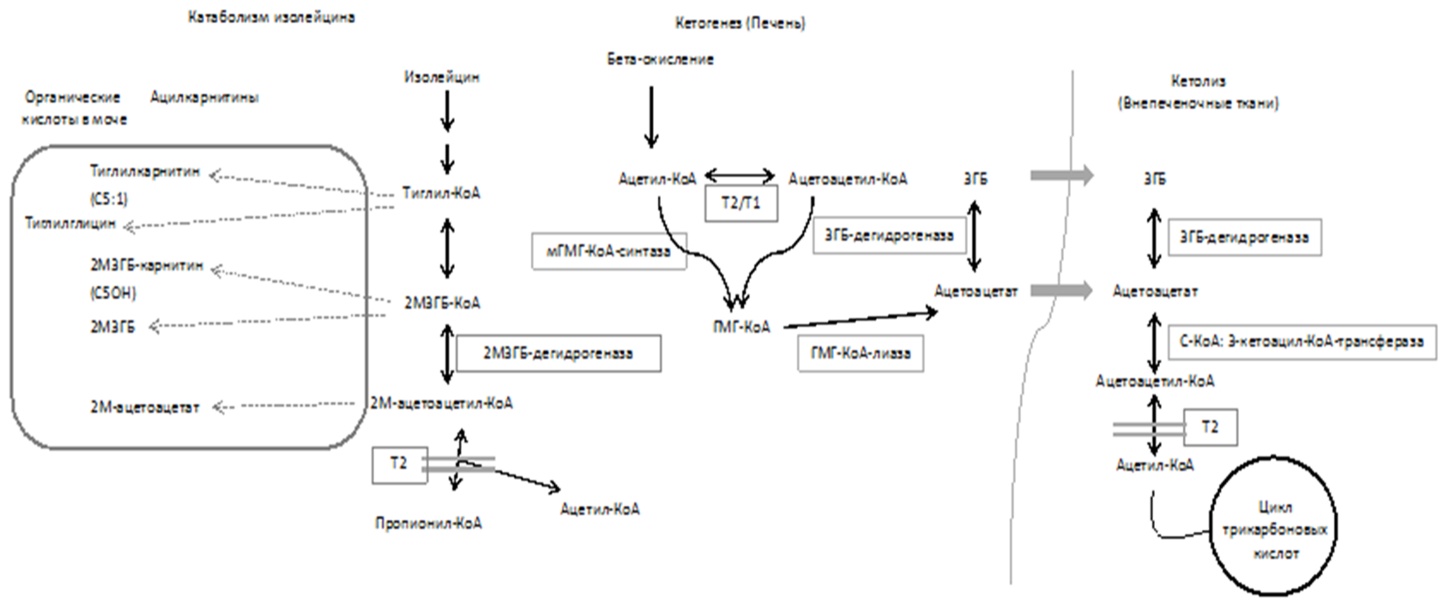
Рисунок 1. Обзор метаболизма кетоновых тел и катаболизм изолейцина.
Т2 (МАТ) участвует в катаболизме изолейцина, кетогенезе в печени и кетолизе во внепеченочных тканях. КоА = кофермент А, 2М3ГБ = 2-метил-3-гидроксибутират, 2М3ГБ-КоА = 2-метил-3-гидроксибутирил-КоА, 2М3ГБ-дегидрогеназа = 2-метил-3-гидроксибутират дегидрогеназа, 2М-ацетоацетат = 2-метилацетоацетат, 2М-ацетоацетил-КоА = 2-метилацетоацетат-КоА, 3ГБ = 3-гидроксибутират, 3ГБ-дегидрогеназа = 3-гидроксибутиратдегидрогеназа, ГМГ-КоА = 3-гидрокси-3-метил-глутарил-КоА, ГМГ-КоА-лиаза = 3-гидрокси-3-метил-глутарил-КоА-лиаза, мГМГ-КоА-синтаза = митохондриальная ГМГ-КоА-синтаза, С-КоА:3-кетоацил-КоА-трансфераза = сукцинил-КоА 3-кетоацил-КоА-трансфераза (SCOT), Т1 = митохондриальная 3-кетоацил-КоА-тиолаза, Т2 = митохондриальная ацетоацетил-КоА-тиолаза (МАТ).
1.4 Особенности кодирования заболевания или состояния (группы заболеваний или состояний) по Международной статистической классификации болезней и проблем, связанных со здоровьем
Согласно МКБ-10, НМАТ относится к классу IV - Болезням эндокринной системы, расстройству питания и нарушению обмена веществ.
МКБ-10: E71.1 - Другие виды нарушения обмена аминокислот с разветвленной цепью.
МКБ-11: 5C50.DY - Другие виды нарушений обмена аминокислот с разветвленной цепью.
OMIM: 203750
ORPHA code: 134
1.5 Классификация заболевания или состояния (группы заболеваний или состояний)
Классификация отсутствует.
1.6 Клиническая картина заболевания или состояния (группы заболеваний или состояний)
Возраст начала НМАТ варьирует от 2 дней до 8 лет. У 80% пациентов заболевание манифестирует в первые два года жизни. Частота манифестации в неонатальном периоде низкая и не превышает 3,5% [4].
НМАТ характеризуется изменением спектра органических кислот в моче в виде повышения концентрации 2М3ГБ, 2-метилацетоацетата и тиглилглицина, а также повышением концентрации ацилкарнитинов крови (C5OH, C5:1). У подавляющего числа пациентов заболевание дебютирует остро с развитием метаболической декомпенсации (криза), тяжелого кетоацидоза, рвоты, обезвоживания, гипотонии, дыхательной недостаточности, судорог и летаргии. В редких случаях заболевание дебютирует с прогрессирующими неврологическими симптомами, такими как мышечная гипотония, судороги, задержка моторного развития, различные гиперкинезы (хорея, миоклонус), атаксия и параплегия [5, 12, 13 - 16].
Метаболический криз обычно характеризуется тяжелым метаболическим ацидозом (pH < 7,0), кетозом, нарушением сознания и комой. Частота эпизодов уменьшается с возрастом и между ними заболевание обычно протекает бессимптомно. Приступы кетоацидоза часто вызваны стрессом: голоданием, острыми заболеваниями (например, гастроэнтерит, респираторные заболевания), лихорадкой, инфекцией и вакцинацией [17]. Во время кетоацидотических кризов при НМАТ изменяется уровень глюкозы в крови, варьируя от 0,6 до 23,3 ммоль/л. Как правило, гипогликемия при кетоацидотических приступах встречается редко [18]. Известен случай генетически подтвержденного НМАТ с множественными эпизодами гипокетотической гипогликемии в неонатальном и детском возрасте [18]. В Японии был также зафиксирован случай кетоацидотического криза при НМАТ без повышения уровня C5OH и C5:1 [19].
У пациентов с НМАТ также может отмечаться умеренная гипераммониемия и повышение печеночных трансаминаз [4]. У некоторых пациентов наблюдается снижение свободного карнитина в плазме крови [15, 20]. В исследовании пациентов с НМАТ разной этнической принадлежности [4] у 19,6% наблюдалась задержка развития (40/204) часто в сочетании с неврологическими нарушениями. Двигательные нарушения встречались в 6,3% случаев (13/204), и включали нарушение мышечного тонуса, хореоатетоз и миоклонические гиперкинезы. Эпилептические судороги вне кризов встречаются достаточно редко [4] и связаны с перенесенным метаболическим кризом. У единичных пациентов возможно развитие кардиомиопатии [21].
При проведении магнитно-резонансной томографии (МРТ) головного мозга обнаруживают повышение Т2 сигнала в области базальных ганглиев с поражением бледного шара, скорлупы, чечевицеобразного и хвостатого ядер и черной субстанции. У пациентов с НМАТ наблюдается поражение наружной и внутренней капсулы, корковая и подкорковая атрофия больших полушарий, вовлечение среднего мозга, а также рассеянные очаги в перивентрикулярном и подкорковом белом веществе головного мозга [4].
2. Диагностика заболевания или состояния (группы заболеваний или состояний) медицинские показания и противопоказания к применению методов диагностики
Обращаем внимание, что, согласно требованиям к разработке клинических рекомендаций, к каждому тезису-рекомендации необходимо указывать силу рекомендаций и доказательную базу в соответствии со шкалами оценки уровня достоверности доказательств (УДД) и уровня убедительности рекомендаций (УУР). Для многих тезисов УУР и УДД будет низким по причине отсутствия посвященных им клинических исследований высокого дизайна. Невзирая на это, они являются необходимыми элементами обследования пациента для установления диагноза и выбора тактики лечения.
Критерии установления диагноза и состояния.
Критерием установления диагноза НМАТ является определение повышенной концентрации уровня 2М3ГБ, тиглилглицина и 2-метилацетоацетата при количественном и качественном анализе органических кислот мочи, а также повышение концентрации ацилкарнитинов крови (C5OH, C5:1).
Диагноз устанавливается на основании совокупности анамнестических данных, клинических данных, результатов лабораторного исследования (биохимического и молекулярно-генетического анализа). Выявление патогенных и вероятно-патогенных вариантов гена ACAT1 по результатам ДНК-диагностики позволяет подтвердить диагноз на молекулярно-генетическом уровне.
2.1 Жалобы и анамнез
При сборе анамнеза и жалоб необходимо обратить внимание на следующие жалобы и анамнестические события:
- отягощенный семейный анамнез (сходные симптомы у родных братьев и сестер пробанда, близкородственный брак);
- манифестация заболевания от 2 дней до 8 лет;
- остановка/задержка/утрата ранее приобретенных психомоторных навыков;
- вялость, слабость;
- угнетение сознания от сонливости до комы;
- судорожные приступы;
- нарушения вскармливания, рвота;
- дыхательные нарушения.
Жалобы и анамнез также описаны в разделе "Клиническая картина".
2.2 Физикальное обследование
При осмотре необходимо обратить внимание на основные клинические проявления НМАТ:
- мышечная гипотония ("синдром вялого ребенка");
- отставание психомоторного развития;
- гепатомегалия;
- судорожные приступы.
2.3 Лабораторные диагностические исследования
Обращаем внимание, что, согласно требованиям к разработке клинических рекомендаций, к каждому тезису-рекомендации необходимо указывать силу рекомендаций и доказательную базу в соответствии со шкалами оценки уровня достоверности доказательств (УДД) и уровня убедительности рекомендаций (УУР). Для многих тезисов УУР и УДД будет низким по причине отсутствия посвященных им клинических исследований высокого дизайна. Невзирая на это, они являются необходимыми элементами обследования пациента для установления диагноза и выбора тактики лечения.
Основные лабораторные методы подтверждения диагноза НМАТ включают количественный и качественный анализ органических кислот мочи, определение ацилкарнитинов крови и молекулярно-генетические исследования гена ACAT1. Данные исследования проводятся в специализированных генетических лабораториях.
- Рекомендуется комплексное определение содержания органических кислот в моче пациентам с клиническими признаками, характерными для НМАТ, с целью подтверждения диагноза биохимическими методами и дифференциальной диагностики с другими наследственными нарушениями обмена веществ [15, 34].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5).
Комментарии: Профили органических кислот в моче при НМАТ характеризуются повышенной экскрецией тиглилглицина, 2М3ГБ и 2-метилацетоацетата как при кетоацидотических кризах, так и в межприступный период.
- Рекомендовано: комплексное определение концентрации на аминокислоты и ацилкарнитины в крови методом тандемной масс-спектрометрии пациентам с клиническими признаками, характерными для НМАТ, с целью подтверждения диагноза биохимическими методами и дифференциальной диагностики с другими наследственными нарушениями обмена веществ [6, 15, 34]
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5).
Комментарии: Характерным маркером НМАТ являются высокие концентрации C5:1 и C5OH. У некоторых пациентов отмечают снижение свободного карнитина [15, 20]. Нормальный уровень данных метаболитов, однако, не позволяет исключить НМАТ: ложноотрицательные результаты при проведении неонатального скрининга неоднократно были зафиксированы в разных лабораториях, которые используют разные отрезные точки. Более того, известен случай кетоацидотического криза при НМАТ с нормальным уровнем C5OH и C5:1 в крови [19]. Алгоритм скрининга при НМАТ приведен в приложении Б.
- Рекомендовано: молекулярно-генетическое исследование мутаций в гене ACAT1 (Комплекс исследований для диагностики органических ацидурий) всем пациентам с биохимическими изменениями, характерными для НМАТ, с целью подтверждения диагноза на молекулярно-генетическом уровне [8, 34]
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5).
Комментарии: Большинство мутаций у пациентов с НМАТ могут быть обнаружены с помощью секвенирования по Сэнгеру всех экзонов и прилегающих к ним участков интронов гена ACAT1, но в небольшом проценте случаев мутацию с применением стандартных методов обнаружить не удается. На сегодняшний день известно по меньшей мере о 105 мутациях в гене ACAT1, большинство которых являются миссенс-мутациями [5]. Так, например, в китайской популяции преобладают патогенные варианты: c.622C>T (p.Arg208Ter), c.1006-1G>C и c.1124A>G (p.Asn375Ser) [6], в индийской - вариант c.578T>G (p.Met193Arg) [7], а во Вьетнаме превалирует патогенный вариант c.622C>T (p.Arg208Ter) [8, 9].
- Рекомендуется исследование уровня водородных ионов (pH) крови; исследование уровня буферных веществ в крови; обнаружение кетоновых тел в моче пациентам с клиническими признаками, характерными для НМАТ, с целью своевременной коррекции терапии при подозрении на развитие метаболического криза [8, 11, 33].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5).
Комментарии: Лабораторными предвестниками развития метаболического криза являются тенденция к метаболическому ацидозу с дефицитом оснований. Основным биохимическим маркером при НМАТ является высокая концентрация кетоновых тел. В период кетоацидотических кризов концентрация кетоновых тел в крови у пациентов с НМАТ превышает 7 ммоль/л [11].
- Рекомендовано исследование уровня глюкозы, аммиака, молочной кислоты в крови пациентам с клиническими признаками, характерными для НМАТ, и при подозрении на метаболический ацидоз с целью диагностики и своевременной коррекции терапии [4, 15, 24, 33, 34].
Уровень убедительности рекомендаций B (уровень достоверности доказательств - 3).
Комментарии: у пациентов с НМАТ может отмечаться умеренная гипераммониемия [4]. Концентрации молочной кислоты (лактата) следует измерять во время первых метаболических кризов у недиагностированных пациентов, чтобы исключить врожденный лактатацидоз [11]. Уровень глюкозы в крови обычно в пределах нормы, но в отдельных случаях может наблюдаться гипогликемия (< 2,5 ммоль/л) или гипергликемия (> 7,8 ммоль/л). Так, уровень глюкозы в крови во время кетоацидотических кризов у пациентов с НМАТ варьирует от 0,6 до 23,3 ммоль/л [18].
- Рекомендуется анализ крови биохимический общетерапевтический всем пациентам с клиническими признаками, характерными для НМАТ, с целью выявления поражения печени [4, 25 - 27].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5).
Комментарии: проводят исследование уровня альбумина, общего белка, общего билирубина, свободного и связанного билирубина, мочевины, креатинина, калия, натрия в крови; исследование уровня железа сыворотки крови; определение активности аспартатаминотрансферазы, аланинаминотрансферазы, гамма-глютамилтрансферазы в крови. У пациентов с НМАТ наблюдается повышение активности трансаминаз [4].
2.4 Инструментальные диагностические исследования
- Рекомендуется компьютерная томография головного мозга и/или магнитно-резонансная томография головного мозга пациентам с клиническими признаками, характерными для НМАТ, с целью оценки состояния головного мозга и дифференциальной диагностики с другими наследственными заболеваниями [4].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 3).
Комментарии: при проведении компьютерной томографии/магнитно-резонансной томографии головного мозга выявляют повышение Т2 сигнала в области базальных ганглиев с поражением бледного шара, скорлупы, чечевицеобразного и хвостатого ядер и черной субстанции. Также наблюдаются поражение наружной и внутренней капсулы, корковая и подкорковая атрофия больших полушарий, вовлечение среднего мозга, а также рассеянные очаги в перивентрикулярном и подкорковом белом веществе головного мозга [4]. Следует отметить, что данные нарушения не являются высокоспецифичными и могут наблюдаться при других заболеваниях. Наркоз проводится по показаниям.
- Рекомендуется электроэнцефалография пациентам с клиническими признаками, характерными для НМАТ, с целью своевременной диагностики эпилепсии и контроля лечения [4, 28, 34].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5).
Комментарии: Эпилептические судороги вне кризов встречаются достаточно редко [4] и связаны с перенесенным метаболическим кризом. Исследования проводят во время диагностики и при диспансерном наблюдении, при необходимости.
- Рекомендуется регистрация электрокардиограммы пациентам с клиническими признаками, характерными для НМАТ, с целью диагностики кардиомиопатии и нарушений ритма сердца [18, 21, 34].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5).
Комментарии: В литературе известны редкие случаи развития кардиомиопатии у пациентов с НМАТ [21].
2.5 Иные диагностические исследования
Для наблюдения пациентов с установленным диагнозом НМАТ необходим мультидисциплинарный подход ввиду того, что данное заболевание характеризуется поражением многих органов и систем, что в свою очередь диктует необходимость совместного ведения пациента специалистами разных профилей. Показаны первичные и повторные консультации врача-генетика, врача-офтальмолога, врача-невролога, врача-гастроэнтеролога, врача-кардиолога, врача-педиатра/врача-терапевта/врача общей практики (семейного врача), а также врачей других специальностей пациентам с НМАТ, имеющим нарушения функций соответствующих органов и систем.
- Рекомендовано: прием (осмотр, консультация) врача-генетика первичный и повторный, при необходимости, всем пациентам с клиническими признаками НМАТ и с установленным диагнозом НМАТ с целью определения тактики ДНК-диагностики, интерпретации полученных результатов, проведения медико-генетического консультирования семье, а также назначения диетотерапии (и далее ее коррекции, при необходимости) [12, 17].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 4).
Комментарии: консультация врача-генетика на этапе диагностики необходима для определения тактики ДНК-диагностики, корректной интерпретации результатов молекулярно-генетических исследований, определения генетических рисков в семье. Интервалы между визитами в случае необходимости назначения/коррекции диетотерапии должны определяться индивидуально с учетом возраста, тяжести заболевания, стабильности обмена веществ и соблюдения диеты.
- Рекомендовано: прием (осмотр, консультация) врача-педиатра первичный и повторный, при необходимости, всем пациентам с НМАТ с целью назначения диетотерапии (и далее ее коррекции при необходимости), а также симптоматического лечения, при необходимости [12, 17].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 4).
Комментарии: Интервалы между визитами должны определяться индивидуально с учетом возраста, тяжести заболевания, стабильности обмена веществ и соблюдения диетотерапии.
- Рекомендовано: прием (осмотр, консультация) врача-невролога первичный и повторный всем пациентам с клиническими признаками, характерными для НМАТ, при подозрении или наличии неврологических нарушений с целью своевременной диагностики и лечения выявленной патологии [4, 5].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 3).
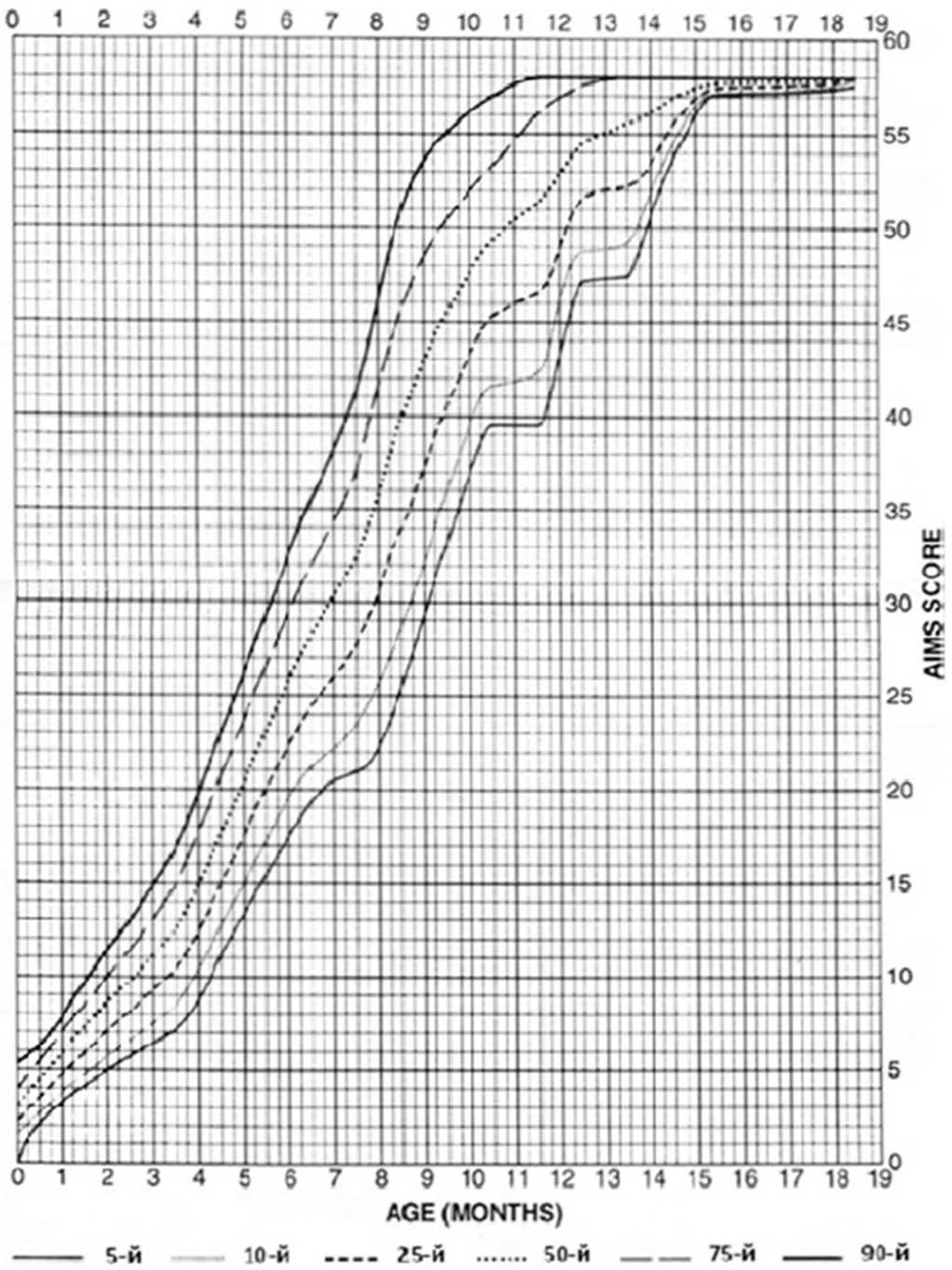
Комментарии: В редких случаях НМАТ дебютирует с прогрессирующими неврологическими симптомами, такими как мышечная гипотония, судороги, задержка моторного развития, различные гиперкинезы (хорея, миоклонус), атаксия и параплегия [5, 12, 13 - 16]. Шкала Альберта моторного развития младенцев приведена в приложении Г1.
- Рекомендовано: прием (тестирование, консультация) медицинского психолога, врача физической и реабилитационной медицины или врача, выполняющего его функцию (специалиста по медицинской реабилитации: врача по лечебной физкультуре и др.) первичный и повторный; разработка индивидуальной программы дефектологической реабилитации; разработка индивидуальной программы логопедической реабилитации пациентам с НМАТ и их семьям с целью оказания психолого-педагогической поддержки [29, 30].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5).
Комментарии: Пациентам необходимо тестирование по утвержденным методикам для определения интеллектуального развития. Психологическая помощь пациентам может потребовать курс занятий в зависимости от индивидуальных особенностей пациента и семьи.
Услуги логопеда:
- Медико-логопедическое исследование при дисфагии;
- Медико-логопедическое исследование при афазии;
- Медико-логопедическое исследование при дизартрии;
- Медико-логопедическая процедура при дисфагии;
- Медико-логопедическая процедура при афазии;
- Медико-логопедическая процедура при дизартрии;
- Медико-логопедическая тонально-ритмическая процедура;
- Медико-логопедическая процедура с использованием интерактивных информационных технологий.
Дифференциальная диагностика.
При кетозе в сочетании с гипогликемией следует проводить дифференциальную диагностику с состояниями, сопровождающимися кетотической гипогликемией, - эндокринными нарушениями (например, дефицит глюкокортикоидов, дефицит гормона роста) и наследственными нарушениями обмена углеводов (например, дефицит гликогенсинтазы, гликогенозы) [15]. Тяжесть кетоацидоза при НМАТ довольно выраженная, что позволяет отличить это расстройство от других причин кетотической гипогликемии. При сочетании кетоацидоза с гипергликемией следует включать в дифференциальную диагностику сахарный диабет и органические ацидемии. При дефиците SCOT (OMIM: 245050) могут наблюдаться кетоацидотические атаки, имитирующие НМАТ. В отличии от НМАТ, для SCOT характерна неонатальная манифестация (50% случаев), постоянный кетоз (даже постпрандиально) и отсутствие характерного для НМАТ профиля органических кислот. При сочетании кетоацидоза с гипераммониемией проводится дифференциальная диагностика с органическими ацидемиями [11].
Пациентам мужского пола также проводится дифференциальная диагностика с дефицитом 2-метил-3-гидроксибутирил-КоА-дегидрогеназы (дефицит 2М3ГБ-КоА дегидрогеназы, OMIM 300438) - редким X-сцепленным нейродегенеративным заболеванием, вызванным патогенными вариантами в гене HSD17B10. Классическая инфантильная форма данного заболевания характеризуется прогрессирующим поражением нервной системы с ретинопатией и кардиомиопатией. Поскольку при дефиците 2М3ГБ-КоА дегидрогеназы задействован тот же метаболический путь катаболизма изолейцина, что и при НМАТ, при анализе ацилкарнитинов у пациентов с этим заболеванием также наблюдается повышение концентрации C5:1 и C5-OH в крови и повышение концентрации 2М3ГБ и тиглилглицина в моче. Единственным отличием является отсутствие 2MAcAc в моче, но иногда из-за его нестабильности он не выявляется даже при НМАТ [11].
Алгоритм дифференциальной диагностики НМАТ с другими заболеваниями, для которых характерен кетоацидотический криз, приведен на рис. 2Б в приложении Б.
3. Лечение, включая медикаментозную и немедикаментозную терапии, диетотерапию, обезболивание, медицинские показания и противопоказания к применению методов лечения
Основные принципы терапии при НМАТ направлены на предотвращение метаболической декомпенсации (метаболического криза), снижение образования токсичных органических кислот, предупреждении развития токсического поражения тканей головного мозга и внутренних органов с обеспечением при этом процессов нормального роста и развития детей.
Основными компонентами лечения пациентов с НМАТ является диетотерапия. По показаниям назначают противоэпилептические препараты и другие виды симптоматической терапии.
3.1 Патогенетическое лечение
- Рекомендуется назначение диетотерапии с ограниченным содержанием белков (0,5 - 1 г/кг/сутки) и жиров, а также избегать длительного голодания пациентам с НМАТ с целью проведения патогенетического лечения [15, 31, 34].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 4).
Комментарии: кетогенная диета противопоказана.
- Рекомендуется увеличить потребление углеводов, а при повышении кетоновых тел в моче проводить инфузии декстрозы** (B05CX другие ирригационные растворы) пациентам с НМАТ, у которых развилась лихорадка и/или рвота, с целью патогенетического лечения и предотвращения метаболического криза [31, 34].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 4).
- Рекомендуется назначение A16AA01 Левокарнитин пациентам с НМАТ, имеющим недостаток свободного карнитина, с целью предотвращения вторичного дефицита карнитина [15, 31, 34].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 4).
Комментарий: начальная доза A16AA01 Левокарнитина в дозировке 50 - 200 мг/кг/сутки, дальнейшая корректировка дозы в зависимости от уровня свободного карнитина. Необходимо регулярное исследование уровня свободного карнитина в крови (комплексное определение концентрации на аминокислоты и ацилкарнитины в крови методом тандемной масс-спектрометрии).
3.1.1 Лечение пациентов в период метаболического криза
При угрозе или в случае развития метаболического криза лечение должно начинаться незамедлительно. Лечебные мероприятия направлены на прекращение образования и накопления токсичных органических соединений и выведение их из организма. Тактика лечения детей в период криза включает коррекцию метаболического ацидоза и водно-электролитных нарушений, а также энергетическую поддержку. При угрозе или в случае развития метаболического криза необходима экстренная госпитализация с целью незамедлительного проведения интенсивной терапии и предотвращения жизнеугрожающих осложнений.
Лечение метаболического криза проводится в отделении реанимации интенсивной терапии врачом-анестезиологом-реаниматологом, а также другими врачами-специалистами (врач-невролог, врач-нефролог, врач-гастроэнтеролог и др.). В период кризов необходимо введение декстрозы** (B05CX другие ирригационные растворы) внутривенно даже при нормальном уровне глюкозы для поддержания ее концентрации на верхней границе нормы, что позволяет подавлять кетогенез. При снижении уровня pH (< 7,1) проводят болюсное введение натрия гидрокарбоната** (B05XA растворы электролитов) (1 ммоль/кг в течение 10 минут) с последующей непрерывной инфузией. При этом следует проводить мониторинг кислотно-основного состояния и электролитов, чтобы избежать гипернатриемии и гипокалиемии. В тяжелых случаях может потребоваться перитонеальный диализ или искусственная вентиляция легких.
3.2 Симптоматическое лечение
- Рекомендовано: прием (осмотр, консультация) врача-невролога первичный и повторный пациентам с диагнозом НМАТ при наличии показаний с целью проведения симптоматической терапии [4].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 3).
Комментарии: не реже 1 раза в 6 месяцев. В редких случаях НМАТ дебютирует с прогрессирующими неврологическими симптомами, такими как мышечная гипотония, судороги, задержка моторного развития, различные гиперкинезы (хорея, миоклонус), атаксия и параплегия [5, 12, 13 - 16].
3.3 Хирургическое лечение
Не применимо.
4. Медицинская реабилитация и санаторно-курортное лечение, медицинские показания и противопоказания к применению методов медицинской реабилитации, в том числе основанных на использовании природных лечебных факторов
Специфической реабилитации пациентам с НМАТ не требуется. В круг реабилитационных мероприятий пациентам с НМАТ могут быть включены занятия психологом/нейропсихологом, логопедом-дефектологом, отдых в специализированных санаториях, а также социальная адаптация с участием специалистов и социальных работников, курсы массажа.
5. Профилактика и диспансерное наблюдение, медицинские показания и противопоказания к применению методов профилактики
5.1 Профилактика
Пациентам с НМАТ следует проводить профилактическую вакцинацию в соответствии с национальным календарем профилактических прививок, включая вакцинацию против гриппа и ротавирусной инфекции, с соблюдением возрастных ограничений препаратов группы J07Вацин на фоне строгого выполнения лечебных назначений с целью формирования эффективного иммунного ответа и предотвращения развития метаболических кризов. Вакцинацию целесообразно проводить под контролем специалистов, имеющих опыт работы с пациентами с метаболическими болезнями. В поствакцинальном периоде следует пристально наблюдать за привитыми с повторным измерением температуры тела. При диагностировании лихорадки > 38,0 °C необходимо незамедлительное назначение жаропонижающей терапии (парацетамол** или ибупрофен**).
В литературе описан случай кетоацидотического криза после вакцинации у ребенка с НМАТ [17].
- Рекомендовано: прием (осмотр, консультация) врача-генетика первичный и повторный, при необходимости, пациенту с диагнозом НМАТ или его официальным представителям с целью проведения медико-генетического консультирования [12].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 4).
Комментарии: семьям с детьми с установленным диагнозом НМАТ необходимо пройти медико-генетическое консультирование с целью определения генетического риска. Как и при других аутосомно-рецессивных заболеваниях, при НМАТ для каждой беременности риск рождения ребенка составляет 25%. Пренатальная диагностика заболеваний, которые поддаются эффективной терапии, возможна, но этически сомнительна. Решение о ее проведении должно быть принято после подробного обсуждения с семьей всех рисков.
5.2 Диспансерное наблюдение
Ежегодно пациенты с установленным диагнозом НМАТ должны проходить углубленную диспансеризацию. Для наблюдения пациентов необходимо применение мультидисциплинарного подхода ввиду того, что данные заболевания характеризуются поражением многих органов и систем, что в свою очередь диктует необходимость совместного ведения пациента специалистами разных профилей. С целью обеспечения комплексной терапии и своевременной ее коррекции при необходимости показаны первичные и повторные консультации врача-генетика, врача-офтальмолога, врача-невролога, врача-гастроэнтеролога, врача-кардиолога, врача-педиатра/врача-терапевта/врача общей практики (семейного врача), а также врачей других специальностей пациентам с НМАТ, имеющим нарушения функций соответствующих органов и систем.
Пациенты с симптоматической эпилепсией нуждаются в наблюдении врача-невролога с опытом работы с пациентами с эпилепсией.
Также осуществляются необходимые реабилитационные мероприятия. Родители должны быть обучены правилам организации терапии в межприступный период и в период угрозы метаболического криза. У ребенка при себе всегда должна находиться памятка с указанием неотложных мероприятий в период начинающегося метаболического криза.
- Рекомендовано: анализ крови биохимический общетерапевтический пациентам с диагнозом НМАТ с целью выявления поражения печени [25 - 27].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5).
Комментарии: Биохимический анализ крови контролируется не реже 1 раза в год. Проводят исследование уровня альбумина, общего белка, общего билирубина, свободного и связанного билирубина, мочевины, креатинина, калия, натрия в крови; исследование уровня железа сыворотки крови; определение активности аспартатаминотрансферазы, аланинаминотрансферазы, гамма-глютамилтрансферазы в крови.
- Рекомендовано: комплексное определение концентрации на аминокислоты и ацилкарнитины в крови методом тандемной масс-спектрометрии пациентам с диагнозом НМАТ с целью контроля лечения [6, 15, 34].
Уровень убедительности рекомендаций B (уровень достоверности доказательств - 3).
Комментарии: Уровни C5OH, C5:1 в крови в результате лечения должны быть приближены к референсным возрастным значениям.
- Рекомендовано: комплексное определение содержания органических кислот в моче пациентам с диагнозом НМАТ с целью своевременного выявления метаболических нарушений и предотвращения развития метаболического криза [6, 15, 34].
Уровень убедительности рекомендаций B (уровень достоверности доказательств - 3).
Комментарии: Кратность проведения анализов зависит от состояния пациента, не реже 1 раза в год.
- Рекомендовано: исследование уровня водородных ионов (pH) крови; исследование уровня буферных веществ в крови; обнаружение кетоновых тел в моче пациентам с диагнозом НМАТ с целью своевременной коррекции терапии при подозрении на развитие метаболического криза [8, 34].
Уровень убедительности рекомендаций B (уровень достоверности доказательств - 3).
Комментарии: Лабораторными предвестниками развития метаболического криза являются тенденция к метаболическому ацидозу с дефицитом оснований. Основным биохимическим маркером при НМАТ является высокая концентрация кетоновых тел. В период кетоацидотических кризов концентрация кетоновых тел в крови у пациентов с НМАТ превышает 7 ммоль/л [11].
- Рекомендовано: диспансерный прием (осмотр, консультация) врача-невролога пациентам с диагнозом НМАТ с целью мониторинга состояния и своевременного выявления патологии нервной системы [4].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 3).
Комментарии: Шкала Альберта моторного развития младенцев приведена в приложении Г1. Оценка проводится 1 раз в 6 месяцев.
- Рекомендовано: диспансерный прием (осмотр, консультация) врача-невролога, врача-генетика, врача-педиатра, прием (осмотр, консультация) врача-диетолога первичный и повторный (при необходимости) пациентам с диагнозом НМАТ с целью назначения диетотерапии (и далее ее коррекции при необходимости) и назначения патогенетической терапии (и далее ее коррекции при необходимости) [4, 12, 17, 32].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 5).
Комментарии: Прием не реже 4 раз в год во время коррекции диетотерапии. Частота осмотра специалиста индивидуальна после подбора диетотерапии. Как правило, после 8 лет посещение специалиста происходит реже в связи с метаболической компенсацией.
6. Организация оказания медицинской помощи
Показания для плановой госпитализации в медицинскую организацию:
1. Проведение диагностики и лечения, требующие продолжительного (дневное или круглосуточное пребывание) медицинского наблюдения и мониторинга клинико-лабораторных показателей;
2. Необходимость проведения различных видов экспертиз или обследования в медицинской организации при невозможности проведения их в амбулаторных условиях (в том числе оформление заключения федерального консилиума/врачебной комиссии).
Показания для экстренной госпитализации в медицинскую организацию:
1. Острые угрожающие для жизни и развития осложнений состояния, требующие неотложного лечения, в том числе интенсивной терапии, а также круглосуточного медицинского наблюдения и проведения специальных видов обследования и лечения.
Показания к выписке пациента из медицинской организации:
1. Отсутствие угрозы для жизни пациента и угрозы развития осложнений, требующих неотложного лечения по завершении диагностических мероприятий.
2. Стабилизация состояния и основных клинико-лабораторных показателей патологического процесса, отсутствие необходимости в продолжительном медицинском наблюдении (дневное или круглосуточное пребывание);
3. Выполнен план обследования и лечения пациента, даны рекомендации по дальнейшему наблюдению и лечению.
4. Необходимость перевода пациента в другое медицинское учреждение или учреждение социального обеспечения.
7. Дополнительная информация (в том числе факторы, влияющие на исход заболевания или состояния)
Прогноз состояния и уровня психического развития пациента зависит от тяжести заболевания, а также сроков начала специализированной терапии.
При своевременной коррекции и профилактике метаболических кризов, соблюдении и строгом контроле патогенетической терапии и симптоматического лечения прогноз для жизни благоприятный. У большого числа пациентов неврологические проявления не развиваются даже после метаболического криза.
Одним из важнейших подходов к ранней диагностике НМАТ является скрининг новорожденных
Критерии оценки качества медицинской помощи
N
Критерии качества
Оценка выполнения
1.
Проведено комплексное определение содержания органических кислот в моче
да/нет
2.
Проведено комплексное определение концентрации на аминокислоты и ацилкарнитины в крови методом тандемной масс-спектрометрии
да/нет
3.
Проведено молекулярно-генетическое исследование мутаций в гене ACAT1 (Комплекс исследований для диагностики органических ацидурий) при установлении диагноза
да/нет
4.
Проведен прием (осмотр, консультация) врача-генетика первичный и повторный
да/нет
5
Проведено исследование уровня глюкозы, аммиака, молочной кислоты в крови
да/нет
Список литературы
1. Wojcik MH, Wierenga KJ, Rodan LH, Sahai I, Ferdinandusse S, Genetti CA, Towne MC, Peake RW, James PM, Beggs AH, Brownstein CA. Beta-ketothiolase deficiency presenting with metabolic stroke after a normal newborn screen in two inpiduals. InJIMD Reports, Volume 39 2017 Jul 20 (pp. 45 - 54). Berlin, Heidelberg: Springer Berlin Heidelberg.
2. Краснопольская К.Д. Наследственные болезни обмена веществ. Справочное пособие для врачей. М. 2005; 364.
3. Daum RS, Mamer OA, Lamm PH, Scriver CR. A" new" disorder of isoleucine catabolism. The Lancet. 1971 Dec 11; 298(7737): 1289 - 90.
4.  SC, Sass JO. 2-methylacetoacetyl-coenzyme A thiolase (beta-ketothiolase) deficiency: one disease-two pathways. Orphanet journal of rare diseases. 2020 Dec; 15: 1 - 7.
SC, Sass JO. 2-methylacetoacetyl-coenzyme A thiolase (beta-ketothiolase) deficiency: one disease-two pathways. Orphanet journal of rare diseases. 2020 Dec; 15: 1 - 7.
5. Abdelkreem E, Harijan RK, Yamaguchi S, Wierenga RK, Fukao T. Mutation update on ACAT1 variants associated with mitochondrial acetoacetyl-CoA thiolase (T2) deficiency. Human mutation. 2019 Oct; 40(10): 1641 - 63.
6. Lin Y, Yang Z, Yang C, Hu H, He H, Niu T, Liu M, Wang D, Sun Y, Shen Y, Li X. C4OH is a potential newborn screening marker-a multicenter retrospective study of patients with beta-ketothiolase deficiency in China. Orphanet Journal of Rare Diseases. 2021 Dec; 16: 1 - 9.
7. Abdelkreem E, Akella RR, Dave U, Sane S, Otsuka H, Sasai H, Aoyama Y, Nakama M, Ohnishi H, Mahmoud S, Abd El Aal M. Clinical and mutational characterizations of ten indian patients with beta-ketothiolase deficiency. JIMD Reports, Volume 35. 2017: 59 - 65
8. Nguyen KN, Abdelkreem E, Colombo R, Hasegawa Y, Can NT, Bui TP, Le HT, Tran MT, Nguyen HT, Trinh HT, Aoyama Y. Characterization and outcome of 41 patients with beta-ketothiolase deficiency: 10 years' experience of a medical center in northern Vietnam. Journal of inherited metabolic disease. 2017 May; 40(3): 395 - 401.
9. Fukao T, Nguyen HT, Nguyen NT, Vu DC, Can NT, Van Pham AT, Nguyen KN, Kobayashi H, Hasegawa Y, Bui TP, Niezen-Koning KE. A common mutation, R208X, identified in Vietnamese patients with mitochondrial acetoacetyl-CoA thiolase (T2) deficiency. Mol Genet Metab. 2010 May; 100(1): 37 - 41.
10. Fukao T, Yamaguchi S, Orii T, Hashimoto T. Molecular basis of ![]() -ketothiolase deficiency: Mutations and polymorphisms in the human mitochondrial acetoacetyl-coenzyme a thiolase gene. Human Mutation. 1995; 5(2): 113 - 20.
-ketothiolase deficiency: Mutations and polymorphisms in the human mitochondrial acetoacetyl-coenzyme a thiolase gene. Human Mutation. 1995; 5(2): 113 - 20.
11. Abdelkreem E, Otsuka H, Sasai H, Aoyama Y, Hori T, Aal MA, Mahmoud S, Fukao T. Beta-ketothiolase deficiency: resolving challenges in diagnosis. Journal of Inborn Errors of Metabolism and Screening. 2019 May 30; 4.
12. Vakili R, Hashemian S. A novel mutation of beta-ketothiolase deficiency: the first report from Iran and review of literature. Iranian Journal of Child Neurology. 2018; 12(3): 113.
13. ![]() D, Bernard G, Fukao T,
D, Bernard G, Fukao T,  JC, Chouinard S, Mitchell GA. A treatable new cause of chorea: Beta-ketothiolase deficiency. Movement Disorders. 2013 Jul; 28(8): 1054 - 6.
JC, Chouinard S, Mitchell GA. A treatable new cause of chorea: Beta-ketothiolase deficiency. Movement Disorders. 2013 Jul; 28(8): 1054 - 6.
14.  C, Apaydin H,
C, Apaydin H,  S, Gibson KM. Delayed-onset dystonia associated with 3-oxothiolase deficiency. Movement Disorders: Official Journal of the Movement Disorder Society. 2001 Mar; 16(2): 372 - 5.
S, Gibson KM. Delayed-onset dystonia associated with 3-oxothiolase deficiency. Movement Disorders: Official Journal of the Movement Disorder Society. 2001 Mar; 16(2): 372 - 5.
15. Fukao T. Beta-ketothiolase deficiency. Orphanet encyclopedia. 2004 Sep.
16.  SC, Schmitt RN, Schlatter SM, Gemperle-Britschgi C,
SC, Schmitt RN, Schlatter SM, Gemperle-Britschgi C, ![]() MC, Berg V,
MC, Berg V, ![]() M, Das AM, Demirkol M, Derks TG,
M, Das AM, Demirkol M, Derks TG,  G. Clinical presentation and outcome in a series of 32 patients with 2-methylacetoacetyl-coenzyme A thiolase (MAT) deficiency. Molecular genetics and metabolism. 2017 Sep 1; 122(1-2): 67 - 75.
G. Clinical presentation and outcome in a series of 32 patients with 2-methylacetoacetyl-coenzyme A thiolase (MAT) deficiency. Molecular genetics and metabolism. 2017 Sep 1; 122(1-2): 67 - 75.
17. Mao S, Yang L, Yin X, Yang J, Huang X. Ketoacidotic crisis after vaccination in a girl with beta-ketothiolase deficiency: a case report. Transl Pediatr. 2021 Feb; 10(2): 459 - 463. doi: 10.21037/tp-20-265. PMID: 33708533; PMCID: PMC7944172.
18. Alijanpour M, Sasai H, Abdelkreem E, Ago Y, Soleimani S, Moslemi L, Yamaguchi S, Rezapour M, Hakimi MT, Matsumoto H, Fukao T. Beta-ketothiolase deficiency: A case with unusual presentation of nonketotic hypoglycemic episodes due to coexistent probable secondary carnitine deficiency. JIMD reports. 2019 Mar; 46(1): 23 - 7.
19. Sarafoglou K, Matern D, Redlinger-Grosse K, Bentler K, Gaviglio A, Harding CO, Rinaldo P. Siblings with mitochondrial acetoacetyl-CoA thiolase deficiency not identified by newborn screening. Pediatrics. 2011 Jul; 128(1): e246 - 50.
20. Henry CG, Strauss AW, Keating JP, Hillman RE. Congestive cardiomyopathy associated with beta-ketothiolase deficiency. J Pediatr. 1981; 99: 754 - 7.
21. Fukao, T., Maruyama, S., Ohura, T., Hasegawa, Y., Toyoshima, M., Haapalainen, A.M., Kuwada, N., Imamura, M., Yuasa, I., Wierenga, R.K. and Yamaguchi, S., 2012. Three Japanese patients with beta-ketothiolase deficiency who share a mutation, c. 431A> C (H144P) in ACAT1: subtle abnormality in urinary organic acid analysis and blood acylcarnitine analysis using tandem mass spectrometry. JIMD Reports-Case and Research Reports, 2011/3, pp. 107 - 115.
22. Pandey R, Singh PM, Garg R, Darlong V, Punj J. Perioperative concerns in a beta-ketothiolase-deficient child. Journal of Anesthesia. 2015 Aug; 29: 647-.
23. Vijayakumary T, Kavinda D. Unexplained Tachypneoa and Severe Metabolic Acidosis in a Three-Month-Old Child: A Rare Presentation of Beta-Ketothiolose Deficiency. Cureus. 2022; 14(2).
24.  G, Durmus-Aydogdu S, Ceylaner S, Sass J. Beta-ketothiolase deficiency: An unusual cause of recurrent ketoacidosis. Turkish Journal of Pediatrics. 2017; 59(4).
G, Durmus-Aydogdu S, Ceylaner S, Sass J. Beta-ketothiolase deficiency: An unusual cause of recurrent ketoacidosis. Turkish Journal of Pediatrics. 2017; 59(4).
25. Baker PR. Pathophysiology of Inherited Metabolic Diseases. In Nutrition Management of Inherited Metabolic Diseases: Lessons from Metabolic University 2022 Jun 15 (pp. 33 - 43). Cham: Springer International Publishing.
26. Prietsch V, Lindner M, Zschocke J, Nyhan WL, Hoffmann GF. Emergency management of inherited metabolic diseases. Journal of inherited metabolic disease. 2002 Nov; 25(7): 531 - 46.
27. del Pilar  M, Bravo SB, Barbosa-Gouveia S, Alvarez JV, Couce ML. Proteomics in Inherited Metabolic Disorders. International Journal of Molecular Sciences. 2022 Dec 1; 23(23): 14744.
M, Bravo SB, Barbosa-Gouveia S, Alvarez JV, Couce ML. Proteomics in Inherited Metabolic Disorders. International Journal of Molecular Sciences. 2022 Dec 1; 23(23): 14744.
28. Ozand PT, Rashed M, Gascon GG, Al Odaib A, Shums A, Nester M, Brismar J. 3-Ketothiolase deficiency: a review and four new patients with neurologic symptoms. Brain and Development. 1994 Nov 1; 16: 38 - 45.
29. Reynolds E, Blanchard S, Jalazo E, Chakraborty P, Bailey Jr DB. Newborn Screening Conditions: Early Intervention and Probability of Developmental Delay. Journal of Developmental & Behavioral Pediatrics. 2022 May 13: 10 - 97.
30. Alliance G. Understanding genetics: a district of Columbia guide for patients and health professionals.
31. Arica V, Arica SG, Dag H, Onur H, Obut ![]() ,
,  S. Beta-ketothiolase deficiency brought with lethargy: case report. Human & experimental toxicology. 2011 Oct; 30(10): 1724 - 7.
S. Beta-ketothiolase deficiency brought with lethargy: case report. Human & experimental toxicology. 2011 Oct; 30(10): 1724 - 7.
32. Анисимова И.В. и др. Методические рекомендации "Метод получения сухого пятна крови на тест-бланк для проведения клинико-лабораторных исследований". 2022: 34.
33. Fukao T, Scriver CR, Kondo N; t2 Collaborative Working Group. The clinical phenotype and outcome of mitochondrial acetoacetyl-CoA thiolase deficiency (beta-ketothiolase or T2 deficiency) in 26 enzymatically proved and mutation-defined patients. Mol Genet Metab. 2001; 72(2): 109 - 114. doi: 10.1006/mgme.2000.3113.
34. Abdelkreem E, Otsuka H, Sasai H, et al. Beta-Ketothiolase Deficiency: Resolving Challenges in Diagnosis. Journal of Inborn Errors of Metabolism and Screening. 2016; 4. doi: 10.1177/2326409816636644
Приложение А1
СОСТАВ
РАБОЧЕЙ ГРУППЫ ПО РАЗРАБОТКЕ И ПЕРЕСМОТРУ
КЛИНИЧЕСКИХ РЕКОМЕНДАЦИЙ
1. Анисимова Инга Вадимовна, к.м.н., заведующая отделом организации медицинской помощи ФГБНУ "Медико-генетический научный центр им. академика Н.П. Бочкова", врач-генетик, член Ассоциации медицинских генетиков.
2. Байдакова Галина Викторовна, к.б.н., заведующая Центром коллективного пользования "Метаболом", в.н.с. лаборатории наследственных болезней обмена ФГБНУ "Медико-генетический научный центр им. академика Н.П. Бочкова", член Российского общества медицинских генетиков
3. Баранов Александр Александрович, академик РАН, профессор, д.м.н.; почетный президент Союз. педиатров России, советник руководителя НИИ педиатрии и охраны здоровья детей НКЦ N 2 ФГБНУ "РНЦХ им акад. Б.В. Петровского", профессор кафедры педиатрии и детской ревматологии ФГАОУ "Первый МГМУ им. И.М. Сеченова" Минздрава России (Сеченовский Университет), главный внештатный специалист педиатр Минздрава России
4. Баранова Полина Владимировна, научный сотрудник ЦКП "Метаболом" ФГБНУ "Медико-генетический научный центр им. академика Н.П. Бочкова".
5. Бушуева Татьяна Владимировна, д.м.н., ведущий научный сотрудник лаборатории питания здорового и больного ребенка ФГАУ НМИЦ здоровья детей МЗ РФ, член Европейского общества детских гастроэнтерологов, гепатологов и нутрициологов (EPSGHAN)
6. Вашакмадзе Нато Джумберовна, д.м.н., руководитель отдела орфанных болезней и профилактики инвалидизирующих заболеваний НИИ педиатрии и охраны здоровья детей НКЦ N 2 ФГБНУ "РНЦХ им акад. Б.В. Петровского", профессор кафедры факультетской педиатрии педиатрического факультета ФГБОУ ВО "РНИМУ им. Н.И. Пирогова" Минздрава России
7. Дегтярева Анна Владимировна, д.м.н., проф., зав. отделом педиатрии Института неонатологии и педиатрии Национального медицинского исследовательского центра акушерства, гинекологии и перинатологии им. акад. В.И. Кулакова, профессор кафедры неонатологии Института здоровья детей Первого Московского государственного медицинского университета им. И.М. Сеченова, член Совета Российского общества неонатологов
8. Захарова Екатерина Юрьевна, д.м.н., заведующая лабораторией наследственных болезней обмена ФГБНУ "Медико-генетический научный центр им. академика Н.П. Бочкова", член Российского общества медицинских генетиков, член европейского общества по изучению наследственных болезней обмена веществ (SSIEM).
9. Какаулина Виктория Сергеевна, врач-невролог, эпилептолог ГБУЗ "Морозовская ДГКБ ДЗМ", член европейского общества по изучению наследственных болезней обмена веществ (SSIEM).
10. Кекеева Татьяна Николаевна, заведующая медико-генетическим отделением ГБУЗ "Морозовская ДГКБ ДЗМ", врач-генетик.
11. Краснощекова Нина Александровна, врач-педиатр, врач-генетик ГБУЗ "Морозовская ДГКБ ДЗМ".
12. Кузенкова Людмила Михайловна, д.м.н., заведующая отделением психоневрологии и психосоматической патологии ФГАУ "Научный медицинский исследовательский центр здоровья детей" МЗ РФ, член Союза педиатров России.
13. Куцев Сергей Иванович, академик РАН, д.м.н., директор ФГБНУ "Медико-генетический научный центр им. Академика Н.П. Бочкова", президент Ассоциации медицинских генетиков России
14. Лобенская Анастасия Юрьевна - заведующий биохимической лабораторией, врач-лабораторный генетик СПб ГКУЗ "Диагностический центр (медико-генетический)".
15. Михайлова Светлана Витальевна, д.м.н., д.м.н., заведующая отделением ФГАОУ "Российская Детская Клиническая Больница" РНИМУ им Н.И. Пирогова.
16. Назаренко Людмила Павловна, профессор, д.м.н., заместитель директора по научной и лечебной работе, руководитель лаборатории наследственной патологии НИИ медицинской генетики, Томского НИМЦ РАН, член Ассоциации медицинских генетиков.
17. Намазова-Баранова Лейла Сеймуровна, академик РАН, профессор, д.м.н., президент Союза педиатров России; паст-президент EPA/UNEPSA; руководитель НИИ педиатрии и охраны здоровья детей НКЦ N 2 ФГБНУ "РНЦХ им акад. Б.В. Петровского", заведующая кафедрой факультетской педиатрии педиатрического факультета ФГБОУ ВО "РНИМУ им. Н.И. Пирогова" Минздрава России, главный внештатный детский специалист по профилактической медицине Минздрава России
18. Николаева Екатерина Александровна, д.м.н., руководитель отдела клинической генетики ОСП "Научно-исследовательский клинический институт педиатрии имени академика Ю.Е. Вельтищева" ФГАОУ ВО РНИМУ имени Н.И. Пирогова, член Российского общества медицинских генетиков, член Ассоциации медицинских генетиков
19. Печатникова Наталья Леонидовна, руководитель Городского Центра орфанных и других редких заболеваний у детей и подростков ГБУЗ "Морозовская ДГКБ ДЗМ".
20. Свиридова В.В. - врач-генетик отдела организации медицинской помощи ФГБНУ "Медико-генетический научный центр им. академика Н.П. Бочкова", м.н.с лаборатории мутагенеза ФГБНУ "Медико-генетический научный центр им. академика Н.П. Бочкова"
21. Селимзянова Лилия Робертовна, к.м.н., заведующая отделом НИИ педиатрии и охраны здоровья детей НКЦ N 2 ФГБНУ "РНЦХ им акад. Б.В. Петровского", доцент кафедры педиатрии и детской ревматологии ФГАОУ "Первый МГМУ им. И.М. Сеченова" Минздрава России (Сеченовский Университет), доцент кафедры факультетской педиатрии педиатрического факультета ФГБОУ ВО "РНИМУ им. Н.И. Пирогова" Минздрава России, член Союза педиатров России
22. Смирнова Ольга Яковлевна, врач-генетик, старший научный сотрудник отдела стандартизации и изучения основ доказательной медицины НКЦ N 2 ФГБНУ "РНЦХ им акад. Б.В. Петровского"
23. Строкова Татьяна Викторовна, д.м.н., профессор РАН, заведующая отделением педиатрической гастроэнтерологии, гепатологии и диетотерапии Клиники ФГБУН "ФИЦ питания и биотехнологии".
24. Субботин Дмитрий Михайлович, врач-генетик ФГБНУ "Медико-генетический научный центр им. академика Н.П. Бочкова", член Ассоциации медицинских генетиков.
25. Сумина Мария Геннадьевна, заведующая отделением медико-генетического консультирования, врач-генетик ГАУЗ Свердловской области "Клинико-диагностический центр "Охрана здоровья матери и ребенка".
26. Таран Наталия Николаевна, к.м.н., старший научный сотрудник отделения педиатрической гастроэнтерологии, гепатологии и диетотерапии ФГБУН "ФИЦ питания и биотехнологии", ассистент кафедры ФГАОУ ВО РНИМУ им. Н.И. Пирогова Минздрава России.
Авторы подтверждают отсутствие финансовой поддержки/конфликта интересов, который необходимо обнародовать.
Приложение А2
МЕТОДОЛОГИЯ РАЗРАБОТКИ КЛИНИЧЕСКИХ РЕКОМЕНДАЦИЙ
Настоящие рекомендации предназначены для применения медицинскими организациями и учреждениями федеральных, территориальных и муниципальных органов управления здравоохранением, систем обязательного и добровольного медицинского страхования, другими медицинскими организациями различных организационно-правовых форм деятельности, направленной на оказание медицинской помощи.
Клинические рекомендации созданы на основании систематического обзора литературы 1971 - 2022 гг. Medline (Pubmed version), Embase (Dialog version) и Cochrane Library databases, с использованием созданных протоколов современных международных клинических рекомендаций по диагностике, лечению и ведению больных с метаболическими болезнями.
НМАТ относится к редким наследственным заболеваниям, что исключает возможность проведения больших когортных и рандомизированных контролируемых исследований и для создания протоколов диагностики и терапии используются лишь тематические исследования экспертов, опубликованные в последние два десятилетия.
Оценка качества доказательств и силы рекомендаций применения медицинских технологий проводилась в соответствии с унифицированной шкалой, приведенной в таблицах 1 - 3.
Целевая аудитория данных клинических рекомендаций:
1. Врачи общей практики (семейные врачи);
2. Врачи-педиатры;
3. Врачи-терапевты;
4. Врачи-генетики;
5. Врачи-лабораторные генетики;
6. Врачи-кардиологи;
7. Врачи-детские кардиологи;
8. Врачи-неврологи;
9. Врачи-гастроэнтерологи;
10. Врачи-рентгенологи;
11. Врачи-офтальмологи;
12. Врачи функциональной диагностики;
13. Медицинские психологи;
14. Студенты медицинских ВУЗов;
15. Обучающиеся в ординатуре и аспирантуре.
Таблица 1. Шкала оценки уровней достоверности доказательств (УДД) для методов диагностики (диагностических вмешательств)
УДД
Расшифровка
1
Систематические обзоры исследований с контролем референсным методом или систематический обзор рандомизированных клинических исследований с применением метаанализа
2
Отдельные исследования с контролем референсным методом или отдельные рандомизированные клинические исследования и систематические обзоры исследований любого дизайна, за исключением рандомизированных клинических исследований, с применением метаанализа
3
Исследования без последовательного контроля референсным методом или исследования с референсным методом, не являющимся независимым от исследуемого метода или нерандомизированные сравнительные исследования, в том числе когортные исследования
4
Несравнительные исследования, описание клинического случая
5
Имеется лишь обоснование механизма действия или мнение экспертов
Таблица 2. Шкала оценки уровней достоверности доказательств (УДД) для методов профилактики, лечения и реабилитации (профилактических, лечебных, реабилитационных вмешательств)
УДД
Расшифровка
1
Систематический обзор РКИ с применением метаанализа
2
Отдельные РКИ и систематические обзоры исследований любого дизайна, за исключением РКИ, с применением метаанализа
3
Нерандомизированные сравнительные исследования, в т.ч. когортные исследования
4
Несравнительные исследования, описание клинического случая или серии случаев, исследования "случай-контроль"
5
Имеется лишь обоснование механизма действия вмешательства (доклинические исследования) или мнение экспертов
Таблица 3. Шкала оценки уровней убедительности рекомендаций (УУР) для методов профилактики, диагностики, лечения и реабилитации (профилактических, диагностических, лечебных, реабилитационных вмешательств)
УУР
Расшифровка
A
Сильная рекомендация (все рассматриваемые критерии эффективности (исходы) являются важными, все исследования имеют высокое или удовлетворительное методологическое качество, их выводы по интересующим исходам являются согласованными)
B
Условная рекомендация (не все рассматриваемые критерии эффективности (исходы) являются важными, не все исследования имеют высокое или удовлетворительное методологическое качество и/или их выводы по интересующим исходам не являются согласованными)
C
Слабая рекомендация (отсутствие доказательств надлежащего качества (все рассматриваемые критерии эффективности (исходы) являются неважными, все исследования имеют низкое методологическое качество и их выводы по интересующим исходам не являются согласованными)
Порядок обновления клинических рекомендаций
Механизм обновления клинических рекомендаций предусматривает их систематическую актуализацию не реже, чем один раз в три года, а также при появлении новых данных с позиции доказательной медицины по вопросам диагностики, лечения, профилактики и реабилитации конкретных заболеваний, наличии обоснованных дополнений/замечаний к ранее утвержденным клиническим рекомендациям, но не чаще 1 раза в 6 месяцев.
Приложение А3
СПРАВОЧНЫЕ МАТЕРИАЛЫ,
ВКЛЮЧАЯ СООТВЕТСТВИЕ ПОКАЗАНИЙ К ПРИМЕНЕНИЮ
И ПРОТИВОПОКАЗАНИЙ, СПОСОБОВ ПРИМЕНЕНИЯ И ДОЗ ЛЕКАРСТВЕННЫХ
ПРЕПАРАТОВ, ИНСТРУКЦИИ ПО ПРИМЕНЕНИЮ
ЛЕКАРСТВЕННОГО ПРЕПАРАТА
1. Федеральный закон от 21.11.2011 N 323-ФЗ (ред. от 28.12.2022) "Об основах охраны здоровья граждан в Российской Федерации" (с изм. и доп., вступ. в силу с 11.01.2023).
2. Приказ Минздрава России от 21.04.2022 N 274н "Об утверждении Порядка оказания медицинской помощи пациентам с врожденными и (или) наследственными заболеваниями".
3. Приказ Минздрава России от 13.10.2017 N 804н (ред. от 24.09.2020, с изм. от 26.10.2022) "Об утверждении номенклатуры медицинских услуг".
4. Приказ Минздрава России от 10.05.2017 N 203н "Об утверждении критериев оценки качества медицинской помощи".
5. Приказ Министерства здравоохранения РФ от 13 октября 2017 г. N 804н "Об утверждении номенклатуры медицинских услуг"
6. Приказ Минздрава России от 07.10.2015 N 700н (ред. от 09.12.2019) "О номенклатуре специальностей специалистов, имеющих высшее медицинское и фармацевтическое образование".
7. Приказ Минздрава России от 28.02.2019 N 103н (ред. от 23.06.2020) "Об утверждении порядка и сроков разработки клинических рекомендаций, их пересмотра, типовой формы клинических рекомендаций и требований к их структуре, составу и научной обоснованности включаемой в клинические рекомендации информации".
8. Информация о лекарственных средствах: https://grls.rosminzdrav.ru/
9. Международная классификация болезней, травм и состояний, влияющих на здоровье (МКБ-10).
Забор биоматериала для диагностики в пятнах крови
Подготовка к взятию крови
Подготовка к взятию крови стандартна: медицинский персонал должен соблюдать правила стерильности, протирая место прокола раствором антисептика (D08A: Антисептики и дезинфицирующие средства), используя одноразовые иглы-скарификаторы и перчатки.
Получение сухого пятна крови
Как правило, для получения сухих пятен [61] кровь берут из пальца, также возможно использование венозной крови, взятой из пробирки с ЭДТА (в зависимости от используемых наборов).
У новорожденных кровь на неонатальный скрининг и ретест берут только из пятки, в исключительных случаях - из пальца (при невозможности взятия крови из пятки).
Место прокола (пятка, палец) следует обработать стерильной салфеткой, смоченной 70% этанолом** после чего промокнуть сухой стерильной салфеткой. Первая капля, образовавшаяся на месте прокола, не используется - ее стирают сухим ватным тампоном. Последующие капли поочередно наносят в круги на впитывающую мембрану тест-бланка. Круги на бланке должны быть пропитаны равномерно, насквозь, без белых пятен на обратной стороне бланка (рис. 1Г). Для получения одного пятна требуется 80 - 100 мкл крови. После нанесения крови на тест-бланк образец выдерживают до полного высыхания в течение не менее 2-х часов при комнатной температуре, не соприкасаясь с другими тест-бланками. После высушивания тест-бланки складываются в индивидуальную упаковку, не соприкасаясь и не накладываясь пятнами крови друг на друга.

- Капли должны полностью пропитать кружки с обратной стороны
- Не касайтесь области, пропитанной кровью

Рисунок 1Г. Сухие пятна крови на тест-бланке.
Для молекулярно-генетических и биохимических исследований не в рамках неонатального скрининга, помимо капельного нанесения, может применяться нанесение необходимого объема цельной крови на тест-бланк полуавтоматическим дозатором, например, из пробирки, содержащей анализируемый биоматериал.
Транспортировка тест-бланков осуществляется при температуре не выше 25 °C [МР СПК].
Алгоритм действий медицинского персонала при взятии образцов крови:
- вымыть руки (гигиенический уровень), надеть перчатки;
- вымыть руки пациента (пятку ребенка в случае, если кровь берется из пятки);
- протереть область прокалывания стерильной салфеткой, смоченной 70% этанолом**, промокнуть сухой стерильной салфеткой; - проколоть стерильным одноразовым скарификатором;
- снять первую каплю крови стерильным сухим тампоном;
- мягко надавить для получения второй капли крови;
- приложить перпендикулярно тест-бланк к капле крови и пропитать его кровью насквозь;
- аналогичным образом нанести на тест-бланк 6 - 8 капель, вид пятен крови должен быть одинаковым с обеих сторон.
- высушить тест-бланк в горизонтальном положении на чистой обезжиренной поверхности не менее 4 ч без применения тепловой обработки и попадания прямых солнечных лучей;
- упаковать тест-бланки в чистый конверт таким образом, чтобы пятна крови не соприкасались.
Особенности при инфузионной терапии
Некоторые пациенты могут получать инфузионную терапию, переливание компонентов крови, что может оказать влияние на результаты тестов. Например, при переливании плазмы крови могут быть получены ложноотрицательные результаты, так как определяемые ферменты находятся в плазме и в клетках крови. Рекомендуется осуществить забор крови для ферментной и ДНК-диагностики не ранее, чем через 6 - 7 дней после переливания плазмы крови и через 7 - 10 дней после переливания компонентов крови.
Не допускается забор крови:
- сразу после проведения пациенту инфузионной терапии;
- сразу после заменного переливания крови.
Хранение и транспортировка биоматериала
Образцы высушенных пятен крови можно хранить в обычной камере холодильника при +4 °C до отправки. Срок хранения до момента отправки не должен превышать 7 дней. Температура при транспортировке биоматериала должна быть от 0 °C до +10 °C. Если хранить дольше и при более высокой температуре, то активность фермента даже в норме может снижаться, что приведет к ложноположительным результатам.
Приложение Б
АЛГОРИТМЫ ДЕЙСТВИЙ ВРАЧА
При повышении концентрации C5:1 при проведении неонатального скрининга необходимо собрать кровь для повторного определения уровня ацилкарнитинов (ретест), если после ретеста уровень C5:1 сохраняется высоким, то образцы отправляют в референсный центр для подтверждающей диагностики. Родителей информируют об особенностях проявления заболевания, сроках проведения тестирования. В референсном центре проводят молекулярно-генетическое исследование и определение органических кислот мочи методом газовой хроматографии с масс-спектрометрией.
В случае отсутствия мутаций в гене ACAT1 и характерного профиля органических кислот мочи необходимо проводить дифференциальную диагностику с другими заболеваниями со сходными биохимическими изменениями (рис. 1Б и рис. 2Б в приложении Б).
При наличии клинических симптомов, что крайне редко для НМАТ в неонатальный период, необходим перевод ребенка в многопрофильный стационар с отделением реанимации и начало терапии в соответствии с клиническими рекомендациями. После подтверждения диагноза необходима медико-генетическая консультация семьи. Дальнейшее ведение пациента осуществляется в соответствии с клиническими рекомендациями.
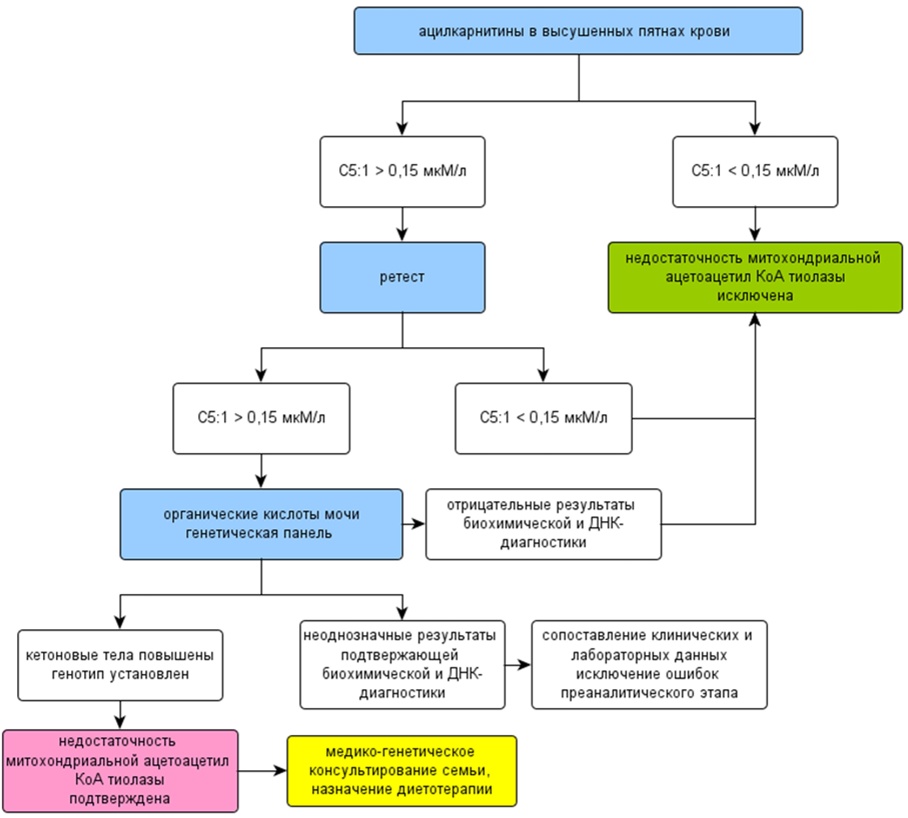
Рисунок 1Б. Алгоритм скрининга на выявление недостаточности митохондриальной ацетоацетил-КоА тиолазы (НМАТ) методом измерения уровня 3-метилкротонилкарнитина (C5:1) в крови и кетоновых тел в моче и молекулярно-генетического исследования гена ACAT1.
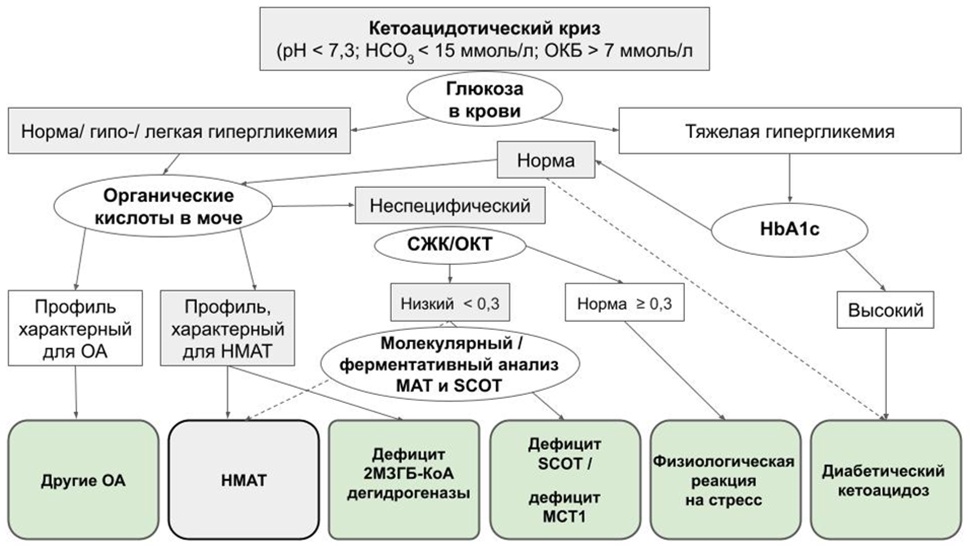
Рисунок 2Б. Алгоритм скрининга на выявление недостаточности митохондриальной ацетоацетил-КоА тиолазы (НМАТ).
2М3ГБ-КоА дегидрогеназа - 2-метил-3-гидроксибутирил-КоА дегидрогеназа; МАТ = митохондриальная ацетоацетил-КоА тиолаза; ОА = органические ацидурии/ацидемии; СЖК/ОКТ = свободные жирные кислоты/общее количество кетоновых тел; HbA1c = гликированный гемоглобин; MCT1 = транспортер монокарбоксилата 1 (monocarboxylate transporter 1); SCOT (succinyl-CoA:3-ketoacid CoA transferase) = сукцинил-КоА 3-кетоацил-КоА-трансфераза (сукцинил КоА:3-оксокислотная КоА-трансфераза).
Отрезная точка (= 0,3) для СЖК/ОКТ не является абсолютной величиной. Длительное голодание может привести к снижению соотношения СЖК/ОКТ. Инфузия декстрозы** снижает уровень СЖК быстрее, чем ОКТ, что также снижает данное соотношение. Тяжелая гипергликемия с повышенным уровнем HbA1c обычно диагностируется как диабетический кетоацидоз, однако известны случаи кетоацидоза схожем с диабетическим при некоторых ОА. Таким образом, нормальный уровень HbA1c не исключает диабетический кетоацидоз (обозначено пунктирной линией). Неспецифический профиль органических кислот в моче при кетоацидозе также не исключает НМАТ (обозначено пунктирной линией). В таком случае рекомендовано проведение молекулярно-генетического исследования или анализа ферментативной активности МАТ и SCOT.
Приложение В
ИНФОРМАЦИЯ ДЛЯ ПАЦИЕНТА
Почему возникает заболевание?
Недостаточность митохондриальной ацетоацетил-КоА тиолазы (НМАТ) - редкое наследственное заболевание, связанное с дефектами (мутациями) в гене ACAT1. Ген находится на 11 хромосоме. Когда в гене ACAT1 происходит мутация, он больше не может служить "инструкцией" для создания фермента МАТ. Таким образом, получившийся фермент функционирует неправильно или же вовсе не производится в организме. НМАТ наследуется по аутосомно-рецессивному типу. Это значит, что у ребенка болезнь проявляется только в том случае, если оба родителя являются носителями болезни и, хотя сами они не болеют, передают ребенку две копии "поврежденного" гена (гена с мутацией). Носитель болезни наследует только одну "поврежденную" копию: либо от отца, либо от матери. Носители не болеют и никаких признаков болезни у них нет. Риск рождения ребенка с НМАТ в семье, где оба родителя являются носителями "поломанного" гена, составляет 25% на каждую беременность (рис. 1В). Мальчики и девочки болеют с одинаковой частотой.
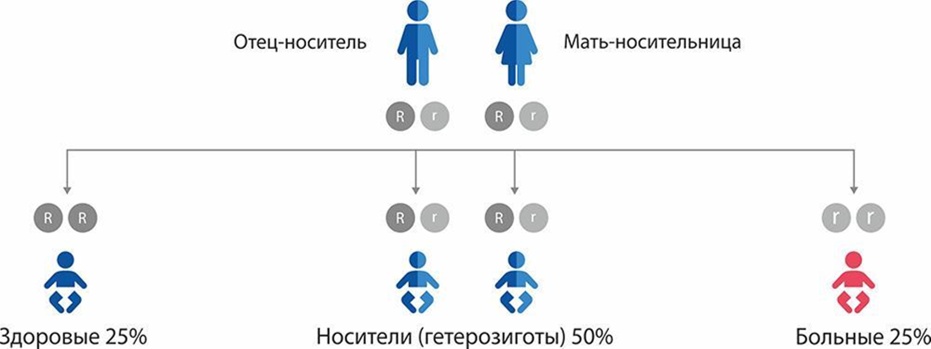
Рисунок 1В. Схема наследования недостаточности митохондриальной ацетоацетил-КоА тиолазы (НМАТ). При наличии двух копий "больного" гена ("r"), унаследованных от обоих родителей-носителей, риск рождения ребенка с НМАТ составляет 25% ("rr").
Что нарушается в метаболизме при НМАТ?
Фермент МАТ играет важную роль в расщеплении белков и жиров в рационе. В частности, МАТ отвечает за переработку изолейцина - аминокислоты, которая входит в состав многих белков. МАТ также перерабатывает кетоны, которые образуются при расщеплении жиров. Если мутация в гене ACAT1 снижает или устраняет активность этого фермента, организм не в состоянии правильно перерабатывать изолейцин и кетоны. В результате могут накапливаться вредные соединения, которые приводят к тому, что кровь становится слишком кислой (кетоацидоз), что ухудшает функцию тканей в особенности центральной нервной системы. Кетоацидоз - патологическое жизнеугрожающее состояние организма, требующее немедленного врачебного вмешательства
Как проявляется НМАТ?
Возраст дебюта НМАТ варьирует от двух дней до восьми лет. В большинстве случаев (80%) заболевание проявляется в возрасте до двух лет.
Заболевание характеризуется эпизодами кетоацидоза (изменение кислотно-щелочного баланса в организме вследствие интенсивного синтеза (производства) кетоновых тел), проявляющимися рвотой, нарушением дыхания, слабостью, сонливостью, которые возникают в младенчестве и обычно прекращаются в подростковом возрасте. Кетоацидоз может привести к коме и летальному исходу при отсутствии лечения. Тем не менее, при своевременной диагностике и соответствующем лечении прогноз для пациентов с НМАТ носит благоприятный характер. Обычно заболевание дебютирует в форме приступа кетоацидоза (кетоацидотического криза), чаще всего вызванного стрессом, голоданием, острыми заболеваниями и/или инфекциями (например, гастроэнтеритом) и в редких случаях повышенным потреблением белка с пищей. О кетоацидозе обычно сигнализирует фруктовый запах изо рта. У ряда пациентов после тяжелых эпизодов развиваются неврологические последствия (например, задержка развития). Редко у пациентов наблюдаются признаки метаболической энцефалопатии (нарушения речи, быстрые, порывистые движения (хорея). В перерывах между эпизодами симптомы у пациентов обычно отсутствуют.
Как устанавливается диагноз?
Первичную диагностику заболевания проводит врач-педиатр. Диагноз ставится на основании клинической картины, анализа мочи (наблюдается высокий уровень 2-метил-3-гидроксимасляной кислоты, 2-метилацетоуксусной кислоты, тиглилглицина и 2-бутанона) и КТ головного мозга. Исследование на НМАТ проводится в рамках ряда программ скрининга новорожденных с помощью тандемной масс-спектрометрии (ТМС) - стандартной методики скрининга новорожденных. Программы скрининга новорожденных доступны в некоторых странах, включая Российскую Федерацию. Для подтверждения диагноза необходимо проведение молекулярно-генетического анализа гена ACAT1.
Как лечат пациентов с НМАТ?
Основные принципы терапии при НМАТ направлены на предотвращение кетоацидоза. Во время кетоацидотического криза врач вводит внутривенно растворы электролитов и раствор декстрозы**. Иногда назначают препараты, содержащие левокарнитин, если у пациентов наблюдается низкий уровень свободного карнитина. Пациентам рекомендуют избегать длительного голодания и жирной (кетогенной) диеты, а также ограничить потребления белка (1,5 - 2 г/кг/сутки).
Как лечат пациентов в период метаболического криза?
Дома при первых признаках заболевания, особенно когда у ребенка снижен аппетит (например, при простуде, гриппоподобных симптомах, вирусных инфекциях, любых заболеваниях, сопряженных с температурой > 37 °C, тонзиллите, гастроэнтерите), следует начать терапию по предотвращению развития криза. Если родители не уверены в появлении у ребенка первых признаков приближающегося заболевания (бледность, сонливость, раздражительность, потеря аппетита, лихорадка, головная боль, ломота и общая боль, кашель, боль в горле или ушах), то в качестве меры предосторожности ребенку следует дать выпить раствор полимера глюкозы - мальтодекстрина - один раз.
Самыми ранними признаками обычно являются незначительные изменения в поведении, которые обычно легко замечают родители. Если ребенок относительно здоров и у него нет рвоты, а только слабость и небольшая сонливость, можно поить его через рот раствором полимера глюкозы частыми дробными порциями (таблица 1В). Точный рецепт/концентрация полимера глюкозы, рекомендованная для каждого ребенка отличается, нужно узнать у лечащего врача что именно подходит вашему ребенку. Некоторым детям не нравится вкус полимера глюкозы, и они предпочитают напитки, которые продаются в супермаркетах, но концентрации углеводов в таких напитках ниже. Покупаемые напитки нужно применять с осторожностью. Низкокалорийные напитки, напитки без добавления сахара содержат очень мало или вовсе не содержат углеводов, поэтому их не следует использовать.
С появлением первых признаков метаболического криза, не дожидаясь прихода врача, следует прекратить прием белка (на 24, максимум на 48 часов), увеличить дозу перорального (через рот) левокарнитина до 50 - 100 мг/кг/сутки и в 2 раза дозу глицина**. Затем в течение 1 - 2 часов нужно регулярно оценивать клиническое состояние ребенка. Если у ребенка постоянная рвота и ему не становится лучше, родителям следует незамедлительно обратиться в больницу для обследования. Врачам скорой помощи необходимо передать выписку с заключением и рекомендациями врача по терапии в период метаболического криза. Родителям нужно взять с собой в больницу всю информацию об использованных растворах и желательно сами растворы.
Таблица 1В. Объем мальтодекстрина при развитии метаболического криза (European registry and network for intoxication type metabolic diseases (E-IMD) 2014).
Возраст (лет)
Мальтодекстрин (%)
Ккал/100 мл
Объем (мл) в день перорально
До 0,5
10
40
Минимально 150 мл/кг
0,5 - 1
12
48
120 мл/кг
1 - 2
15
60
100 мл/кг
2 - 6
20
80
1200 - 1500 мл в сутки
6 - 10
20
80
1500 - 2000 мл в сутки
Старше 10
25
100
2000 - 2500 мл в сутки
Какой нужно проводить мониторинг для пациентов с НМАТ?
Необходим мультидисциплинарный подход к наблюдению и лечению пациентов с НМКК специалистами разных профилей с целью обеспечения комплексной терапии и своевременной ее коррекции при необходимости. Для пациентов с НМАТ необходимы регулярные обследования. В таблице 2В приведен примерный перечень и регулярность исследований, однако точный план обследования согласовывается индивидуально со своим лечащим врачом.
Таблица 2В. Регулярность обследований у пациентов с НМАТ.
Исследование
Интервал
Коррекция диетотерапии (проводят врачи-генетики или врачи-диетологи)
Не реже 1 р/мес на 1 году жизни, далее 1 р/3 мес или по показаниям до 3 лет
Ультразвуковое исследование (УЗИ) органов брюшной полости
В среднем, 1 - 2 раз в год
Проведение электроэнцефалографии (ЭЭГ)
В среднем, 1 - 2 раз в год
Проведение биохимического анализа крови общетерапевтического
Каждые 6 месяцев пациентам до 6 лет, каждые 6 - 12 месяцев пациентам старше 6 лет. Но в период инфекционных заболеваний, при угрозе метаболического криза - не реже 1 раза в 7 - 10 дней.
Общий (клинический) анализ крови
Не реже 1 - 2 раза в год
Исследование уровня свободного карнитина и ацилкарнитинов в крови, комплексное определение содержания органических кислот в моче, комплексное определение содержания органических кислот в моче методом тандемной масс-спектрометрии (ТМС), комплекс исследований для диагностики органических ацидурий, исследование аминокислот и метаболитов в моче
Каждые 6 месяцев пациентам до 6 лет, каждые 6 - 12 месяцев пациентам старше 6 лет. Но в период инфекционных заболеваний, при угрозе метаболического криза - не реже 1 раза в 7 - 10 дней частота исследований в динамике зависит от возраста: чаще у детей до 1 года (ежемесячно) и детей раннего и дошкольного возраста.
Кардиологическое обследование
(консультация врача-кардиолога/врача-детского кардиолога, ЭКГ, ЭХО-КГ)
Не реже 1 - 2 раза в год
Как пациенты получают лечение в Российской Федерации?
НМКК относится к числу редких наследственных болезней обмена веществ и входит в перечень редких (орфанных) заболеваний, лечение которых проводится за счет средств регионального бюджета. После установления диагноза необходимо включение пациента в региональный сегмент регистра по жизнеугрожающим редким (орфанным) заболеваниям, с целью дальнейшего обеспечения необходимым лечебным питанием и лекарственными препаратами. В разных регионах за ведение регистра отвечают разные специалисты, но чаще всего это врачи-генетики, поэтому после установления диагноза обязательно нужно обратиться к региональному врачу-генетику.
Как могут помогать родные и близкие?
Не забывайте: от семьи также зависит успех лечения. Нужно соблюдать рекомендации, ни при каких условиях не допускать "срывов" в диетотерапии, следует уделять внимание реабилитации и плановым обследованиям. Все члены семьи должны знать, что ребенок нуждается в особом питании и близкие родственники должны освоить навыки расчета диеты. Родители пациента с НМКК, а в дальнейшем и сам пациент, должны быть обучены правилам организации терапии в межприступный период и в период угрозы метаболического криза.
У ребенка при себе всегда должна находиться памятка с указанием неотложных мероприятий в период начинающегося метаболического криза.
Приложение Г1 - ГN
ШКАЛЫ ОЦЕНКИ, ВОПРОСНИКИ И ДРУГИЕ ОЦЕНОЧНЫЕ ИНСТРУМЕНТЫ
СОСТОЯНИЯ ПАЦИЕНТА, ПРИВЕДЕННЫЕ В КЛИНИЧЕСКИХ РЕКОМЕНДАЦИЯХ
Приложение Г1
ШКАЛА АЛЬБЕРТА МОТОРНОГО РАЗВИТИЯ МЛАДЕНЦЕВ
Название на русском языке: шкала Альберта моторного развития младенцев.
Оригинальное название: Alberta Infant Motor Scale (AIMS).
Источник (официальный сайт разработчиков, публикация с валидацией): Piper MC, Pinnell LE, Darrah J, Maguire T, Byrne PJ. Construction and validation of the Alberta Infant Motor Scale (AIMS). Can J Public Health. 1992; 83: S46 - 50.
Тип (подчеркнуть):
- шкала оценки
Назначение: моторная шкала для младенцев Альберта (AIMS), шкала наблюдательной оценки, была разработана для измерения общего двигательного созревания у детей от рождения до самостоятельной ходьбы.
Содержание (шаблон): основываясь на литературе, 58 навыков были сгенерированы и распределены по четырем позициям: лежа на спине, лежа на животе, сидя и стоя. Каждому из навыков присваивается определенный балл.
Ключ (интерпретация): баллы суммируются, проводится общая оценка моторного развития и отклонение/соответствие нормальному развитию в перцентилях
Шкала Альберта моторного развития младенцев
число/месяц/год
Фамилия, имя
__________________
Дата исследования:
____/____/___
Идентификационный номер
__________________
Дата рождения:
____/____/___
Исследователь
__________________
Хронологический возраст:
____/____/___
Место проведения исследования
__________________
Скорректированный возраст:
____/____/___
Предшествующие засчитанные пункты
Пункты, засчитанные в "окне"
Балл по шкале
Положение на животе
Положение на спине
Посадка
Вставание
Общий балл: ______ перцентиль: ______
Примечания/рекомендации:
ШКАЛА ОЦЕНКИ МОТОРНОГО РАЗВИТИЯ У МЛАДЕНЦЕВ
N исследования

Положение лежа на животе (1)
Физиологическая флексия. Поворачивает голову для отведения, носа от поверхности.
Положение на животе

Положение лежа на животе (2)
Симметрично поднимает голову под углом 45°. Не способен удерживать голову на одной линии с туловищем.

Положение на животе с опорой
Локти находятся позади плеч. Без поддержки поднимает голову под углом 45°.
Опора на предплечья (1)
Поднимает и удерживает голову под углом более 45°. Локти на одной линии с плечами. Грудная клетка размещается по центру.
Подвижность в положении на животе
Поднимает голову под углом до 90°. Неконтролируемые попытки переноса веса.
Опора на предплечья (2)
Локти находятся перед плечами. Активное подтягивание подбородка с вытягиванием шеи.
Положение на спине
Положение лежа на спине (1)
Физиологическая флексия. Поворот головы; подносит рот к руке. Беспорядочные движения руками и ногами.

Положение лежа на спине (2)
Поворот головы к средней линии. Необязательный асимметричный шейный тонический рефлекс (АШТР).

Положение лежа на спине (3)
Голова на одной линии с туловищем. Двигает руками, но не способен поднести руки к средней линии.
Положение лежа на спине (4)
Активность сгибателей шеи с подтягиванием подбородка. Подносит руки к средней линии.
Поднесение рук к коленям
Подтягивание подбородка. Достает руками до коленей. Активность брюшных мышц.
Посадка
Посадка с поддержкой
Поднимает и кратковременно удерживает голову на одной линии с туловищем.
Посадка с опорой на руки
Удерживает голову на одной линии с туловищем. Кратковременно опирается на руки.
Подтягивание в сидячее положение
Подтягивает подбородок; голова на одной линии с туловищем или впереди.
Вставание
Опора на ноги с поддержкой (1)
Возможно попеременное сгибание в тазобедренном и коленном суставах.
Опора на ноги с поддержкой (2)
Голова на одной линии с туловищем. Бедра находятся позади плеч. Разнообразные движения ногами.
Опора на вытянутые руки
Перекатывание из положения на животе в положение на спине без поворота
Плавание
Руки вытянуты. Подтягивание подбородка с подъемом грудной клетки. Боковой перенос веса.
Движение инициируется головой. Туловище движется как одно целое.
Паттерн активности разгибателей.
Попытка дотянуться до предмета из упора на предплечья
Переворот
Перекатывание из положения на животе в положение на спине с поворотом
Активный перенос веса с одной стороны. Контролируемая попытка дотянуться до предмета свободной рукой.
Перевороты. Движения рук и ног. Боковая флексия туловища.
Поворот туловища
Положение на четвереньках (1)
Поднесение рук к ступням
Перекатывание из положения на спине в положение на животе без поворота
Ноги согнуты. отведены и повернуты кнаружи. Поясничный лордоз. Удерживает положение.
Способен удерживать ноги в среднем положении. Присутствует подвижность в тазовом отделе.
Боковое выпрямление головы. Туловище движется как одно целое.
Перекатывание из положения на спине в положение на животе с поворотом
Активное вытягивание
Посадка без поддержки

Поворот туловища.
Вытягивается, отталкиваясь ногами.
Приведение лопаток и разгибание плеч. Не способен удержать положение.
Посадка с опорой на руки
Посадка без поддержки без опоры на руки
Перенос веса в положении сидя без поддержки
Вытягивание грудного отдела позвоночника. Движения головы, независимые от туловища; опирается на вытянутые руки.
Не может оставаться один в сидячем положении неограниченное время.
Перенос веса вперед, назад или в стороны. Не может оставаться один в сидячем положении.
Посадка без опоры на руки (1)
Попытка дотянуться до предмета с поворотом в положении сидя
Вставание с поддержкой (3)

Руки не прижаты к телу. Способен играть с игрушкой. Может оставаться один в сидячем положении.
Сидит самостоятельно. Тянется к игрушке с поворотом туловища.
Бедра находятся на линии плеч. Активный контроль туловища.
Разнообразные движения ногами
Положение лежа на боку с опорой
Реципрокное ползание
Перемещение из положения на четвереньках в положение сидя или полусидя
Разобщенность ног. Неподвижность плеч. Поворот в пределах оси тела.
Реципрокные движения рук и ног с поворотом туловища.
Перемещается в данное положение и из него. Может принимать сидячее положение.
Реципрокное ползание на четвереньках (1)
Попытка дотянуться до предмета из упора на вытянутую руку
Положение на четвереньках (2)
Ноги отведены и повернуты кнаружи. Поясничный лордоз; перенос веса с одной стороны на другую с боковой флексией туловища.
Тянется к предмету вытянутой рукой. Поворот туловища.
Бедра выровнены под тазом. Уплощение поясничного отдела.
Модифицированное положение на четвереньках
Перемещение из положения сидя в положение на животе
Перемещение из положения сидя на четвереньки
Перемещается в данное положение. Возможно продвижение вперед.
Перемещается из положения сидя, чтобы лечь на живот. Подтягивается руками, ноги неактивны.
Активно поднимает таз, ягодицы и ненагруженную ногу, чтобы встать на четвереньки.
Самостоятельное стояние
Первые шаги
Вставание из модифицированного положения на корточках


В течение короткого времени стоит самостоятельно. Уравновешивает реакции в ступнях.
Ходит самостоятельно; передвигается быстро мелкими шагами.
Перемещается из положения на корточках в положение стоя с контролируемой флексией и выпрямлением бедер и коленей.
Вставание из опоры на четыре конечности
Самостоятельное хождение
Сидение на корточках
![]()
Быстро отталкивается ладонями, чтобы встать.
Ходит самостоятельно.
Сохраняет положение, уравновешивая реакции в ступнях и положение туловища.
Посадка без опоры на руки (2)
Подтягивается для вставания с опорой
Подтягивается для вставания/стоит

Положение ног варьирует. Младенец легко перемещается из одного положения в другое.
Отталкивается руками и выпрямляет колени.
Подтягивается, чтобы встать; переносит вес с одной стороны на другую
Вставание с опорой с поворотом
Хождение с опорой без поворота
Положение на одном колене

Поворот туловища и таза.
Ходит с опорой в боковом направлении без поворота.
Может вставать или перемещаться в данное положение.
Контролируемое опускание из положения стоя
Реципрокное ползание на четвереньках (2)
Хождение с опорой с поворотом


Контролируемое опускание из положения стоя.
Плоский поясничный отдел. Движется с поворотом туловища.
Ходит с опорой с поворотом.
Величины перцентилей