Рекомендация Коллегии Евразийской экономической комиссии от 06.08.2019 N 23
КОЛЛЕГИЯ ЕВРАЗИЙСКОЙ ЭКОНОМИЧЕСКОЙ КОМИССИИ
РЕКОМЕНДАЦИЯ
от 6 августа 2019 г. N 23
О РУКОВОДСТВЕ
ПО ОЦЕНКЕ И КОНТРОЛЮ ДНК-РЕАКТИВНЫХ (МУТАГЕННЫХ)
ПРИМЕСЕЙ В ЛЕКАРСТВЕННЫХ СРЕДСТВАХ И УСТАНОВЛЕНИЮ
ГРАНИЦ ПОТЕНЦИАЛЬНОГО КАНЦЕРОГЕННОГО РИСКА
Коллегия Евразийской экономической комиссии в соответствии со статьей 30 Договора о Евразийском экономическом союзе от 29 мая 2014 года и пунктом 3 статьи 3 Соглашения о единых принципах и правилах обращения лекарственных средств в рамках Евразийского экономического союза от 23 декабря 2014 года, а также в целях обеспечения применения единых подходов к оценке безопасности лекарственных препаратов и недопущения обращения на таможенной территории Евразийского экономического союза потенциально опасных лекарственных препаратов
рекомендует государствам - членам Евразийского экономического союза применять Руководство по оценке и контролю ДНК-реактивных (мутагенных) примесей в лекарственных средствах и установлению границ потенциального канцерогенного риска согласно приложению:
по истечении 18 месяцев с даты опубликования настоящей Рекомендации на официальном сайте Евразийского экономического союза - при фармацевтической разработке лекарственных средств и оценке безопасности лекарственных препаратов;
по истечении 36 месяцев с даты опубликования настоящей Рекомендации на официальном сайте Евразийского экономического союза - при назначении в отношении новых лекарственных препаратов клинических исследований, не включающих в себя фазы IIb и III.
При этом исходить из того, что в случае, если реализация положений указанного Руководства осуществляется в соответствии с актами Евразийской экономической комиссии, применение таких положений осуществляется с даты вступления в силу соответствующих актов.
Председатель Коллегии
Евразийской экономической комиссии
Т.САРКИСЯН
Приложение
к Рекомендации Коллегии Евразийской
экономической комиссии
от 6 августа 2019 г. N 23
РУКОВОДСТВО
ПО ОЦЕНКЕ И КОНТРОЛЮ ДНК-РЕАКТИВНЫХ (МУТАГЕННЫХ)
ПРИМЕСЕЙ В ЛЕКАРСТВЕННЫХ СРЕДСТВАХ И УСТАНОВЛЕНИЮ
ГРАНИЦ ПОТЕНЦИАЛЬНОГО КАНЦЕРОГЕННОГО РИСКА
I. Общие положения
1. Синтез активных фармацевтических субстанций (действующих веществ) предполагает применение химически активных веществ, реагентов, растворителей, катализаторов и других технологических добавок. В результате химического синтеза или последующей деградации во всех фармацевтических субстанциях и соответствующих лекарственных препаратах образуются ДНК-реактивные (мутагенные) примеси (далее - мутагенные примеси), способные изменять генетический материал человека и других организмов, что можно установить в соответствующем тесте (например, в тесте на оценку генных мутаций у бактерий). Целью настоящего Руководства является обеспечение применения единой методики идентификации, категоризации, квалификации и контроля мутагенных примесей в составе фармацевтических субстанций и лекарственных препаратов для ограничения создаваемого ими потенциального канцерогенного риска.
2. Настоящее Руководство применяется с учетом правил по изучению примесей в лекарственных средствах и установлению требований к ним в спецификациях и руководства по доклиническим исследованиям безопасности в целях проведения клинических исследований и регистрации лекарственных препаратов, определяемых Евразийской экономической комиссией.
3. В настоящем Руководстве рассматриваются вопросы управления рисками, безопасности и качества лекарственного препарата при установлении уровней содержания мутагенных примесей, которые обеспечивают статистически незначимый уровень канцерогенного риска.
4. Настоящее Руководство включает в себя рекомендации по оценке и контролю мутагенных примесей, которые содержатся или с достаточной долей вероятности могут содержаться в фармацевтической субстанции или лекарственном препарате, с учетом возможного влияния таких примесей на организм человека исходя из предполагаемого медицинского применения лекарственного препарата.
5. Настоящее Руководство распространяется на:
а) новые фармацевтические субстанции и новые лекарственные препараты, находящиеся на этапе клинической разработки или в процессе регистрации;
б) известные фармацевтические субстанции, вводимые в состав лекарственного препарата в процессе его разработки или в пострегистрационный период в следующих случаях:
изменение синтеза известной фармацевтической субстанции приводит к образованию новых примесей или требует ужесточения критериев приемлемости существующих примесей;
изменение производственной рецептуры (состава или технологического процесса производства) приводит к образованию новых продуктов деградации или к необходимости ужесточения критериев приемлемости для существующих продуктов деградации;
изменение показания к применению или режима дозирования лекарственного препарата приводит к существенному изменению допустимого уровня канцерогенного риска.
6. Оценка мутагенного потенциала примесей, предусмотренная настоящим Руководством, не проводится в отношении следующих видов фармацевтических субстанций и групп лекарственных препаратов:
а) биологические (биотехнологические);
б) пептидные;
в) олигонуклеотидные;
г) радиофармацевтические;
д) продукты ферментации;
е) лекарственное растительное сырье, растительные фармацевтические субстанции (препараты на основе лекарственного растительного сырья), лекарственные растительные препараты и неочищенные препараты животного и растительного происхождения.
7. Настоящее Руководство не распространяется на фармацевтические субстанции и лекарственные препараты, предназначенные для лечения распространенного рака. В некоторых случаях фармацевтическая субстанция, предназначенная для применения в онкологии, сама по себе мутагенна при применении в терапевтических концентрациях и может приводить к повышенному канцерогенному риску. При этом экспозиция мутагенной примеси в составе фармацевтической субстанции может не увеличивать существенно канцерогенный риск от применения этой фармацевтической субстанции. Контроль уровня приемлемого содержания мутагенных примесей осуществляется в соответствии с допустимыми пределами для немутагенных примесей.
8. Оценка мутагенного потенциала примесей не проводится в отношении вспомогательных веществ, используемых в зарегистрированных лекарственных препаратах, вкусовых добавках, красителях и ароматизаторах. Настоящее Руководство не распространяется на вещества, экстрагируемые из упаковки лекарственного препарата, однако его можно применять для оценки безопасности с целью ограничения потенциального канцерогенного риска. Принципы оценки такого риска, предусмотренные настоящим Руководством, при необходимости можно использовать в отношении примесей во вспомогательных веществах, используемых впервые в составе лекарственного препарата и являющихся продуктами химического синтеза.
9. Настоящее Руководство содержит указания к нормированию уровня содержания мутагенных примесей, которые даже в малых количествах способны оказывать прямое повреждающее действие на ДНК, приводя к мутациям и к возможному развитию онкологических заболеваний. Такой тип мутагенных примесей, как правило, обнаруживается в тесте обратных мутаций у бактерий (анализ мутагенности). Другие типы генотоксикантов, которые не являются мутагенными, как правило, имеют пороговый характер проявления своего действия и обычно не несут канцерогенный риск для человека при условии, что их содержание не превышает норм, установленных для таких групп примесей. В связи с этим в целях ограничения возможного канцерогенного риска у человека, обусловленного экспозицией потенциальных мутагенных примесей, проводятся испытания на мутагенность у бактерий (тест Эймса), чтобы оценить мутагенный потенциал и необходимость контроля уровня содержания этих примесей.
II. Определения
10. Для целей настоящего Руководства используются понятия, которые означают следующее:
"генотоксичность" (genotoxicity) - вредное изменение генетического материала независимо от механизма происхождения этого изменения;
"ДНК-реактивность" (DNA-reactive) - способность вызывать прямое повреждение ДНК путем химического взаимодействия с ней;
"допустимое поступление" (acceptable intake) - уровень поступления примеси, несущий ничтожно низкий канцерогенный риск или в случае тяжелых (угрожающих жизни) заболеваний уровень, оправданный допустимым соотношением "польза - риск";
"допустимый предел" (acceptable limit) - максимальная допустимая концентрация примеси в фармацевтической субстанции или лекарственном препарате, рассчитанная на основе допустимого поступления и суточной дозы препарата;
"количественная структурно-функциональная зависимость, (quantitative structure-activity relationship, QSAR) - зависимость между структурой соединения или фрагментом (функциональной группой) соединения и мутагенной активностью этой структуры (фрагмента), установленная на основе анализа экспериментальных данных;
"коэффициент очистки" (purge factor) - отношение уровня содержания примеси в начале процесса к уровню ее содержания на конечном этапе процесса, который рассчитывается на основе непосредственного измерения или прогнозирования;
"кумулятивное поступление" (cumulative intake) - суммарное поступление вещества, действию которого подвергается человек в течение длительного времени;
"мутагенная примесь" (mutagenic impurity) - примесь, проявившая способность изменять генетический материал в соответствующем тесте, например в тесте на оценку генных мутаций у бактерий;
"структурный признак" (structural alert) - химическая группа или фрагмент молекулы (молекулярная субструктура), для которых установлена или возможна связь с мутагенным действием;
"ТД50" - доза, приводящая к 50-процентной частоте развития онкологического заболевания, что эквивалентно вероятности канцерогенного риска, равного 1 к 2;
"экспертное знание" - анализ имеющихся данных и использование других значимых сведений для оценки точности прогноза мутагенности с помощью компьютерного моделирования.
Иные понятия, используемые в настоящем Руководстве, применяются в значениях, определенных Руководством по составлению нормативного документа по качеству лекарственного препарата, утвержденным Решением Коллегии Евразийской экономической комиссии от 7 сентября 2018 г. N 151, Руководством по валидации процесса производства лекарственных препаратов для медицинского применения (приложение к Рекомендации Коллегии Евразийской экономической комиссии от 26 сентября 2017 г. N 19), Руководством по качеству лекарственных растительных препаратов (приложение к Рекомендации Коллегии Евразийской экономической комиссии от 10 мая 2018 г. N 6), Руководством по выбору тестов и критериев приемлемости для составления спецификаций на лекарственное растительное сырье, растительные фармацевтические субстанции (препараты на основе лекарственного растительного сырья) и лекарственные растительные препараты (приложение к Рекомендации Коллегии Евразийской экономической комиссии от 12 февраля 2019 г. N 6).
III. Методология оценки и контроля мутагенных примесей
11. Оценка структуры примеси позволяет прогнозировать ее мутагенность в бактериальных тест-системах на основании имеющихся знаний. Такая оценка может проводиться путем анализа научной литературы и (или) компьютерного моделирования токсикологической оценки.
12. Порог токсикологической угрозы определяет общее допустимое поступление неизученного химического соединения, которое вызывает ничтожно низкий канцерогенный риск или другие токсические эффекты. Расчет порога токсикологической угрозы следует выполнять консервативными методами с использованием:
простой линейной экстраполяции дозы (ТД50);
величины экстраполяции дозы (ТД50), установленной для наиболее чувствительных видов животных и наиболее чувствительных зон развития опухолей.
Порог токсикологической угрозы при таком расчете представляет собой дозу, приводящую к развитию опухолей у человека с частотой 1 случай на 1 000 000. В случае присутствия мутагенных примесей в фармацевтических субстанциях и лекарственных препаратах допускается использовать в качестве значения величины порога токсикологической угрозы эмпирическую величину дозы 1,5 мкг/сут, что соответствует теоретическому повышению пожизненного риска возникновения опухолей у человека с частотой 1 случай на 100 000. Для ряда функциональных групп в молекулах химических соединений установлена настолько высокая мутагенная активность, что даже при поступлении в организм человека в меньших дозах, чем порог токсикологической угрозы, будет возникать теоретическая вероятность существенного канцерогенного риска. Такие высокоактивные мутагенные канцерогены, называются "группой вызывающей опасения", и включают в себя афлатоксиноподобные, N-нитрозосоединения и алкил-азоксисоединения.
13. На ранних фазах клинической разработки лекарственного препарата ожидается, что стратегии контроля и подходы к безопасности в недостаточной степени разработаны, поскольку накоплен малый объем сведений о препарате. В целях настоящего Руководства допустимое поступление мутагенных примесей основывается на выработанных стратегиях оценки рисков. Допустимый риск в ранней фазе разработки устанавливается на теоретически рассчитанном уровне содержания мутагенных примесей, приводящем к возникновению опухолей у человека приблизительно с частотой 1 случай на 1 000 000. Допустимое повышение канцерогенного риска для новых лекарственных препаратов на более поздних этапах разработки и для зарегистрированных лекарственных препаратов устанавливается на теоретически рассчитанном уровне возникновения опухолей у человека с частотой 1 случай на 100 000. Такие уровни риска позволяют теоретически обеспечить незначительное повышение канцерогенного риска на протяжении жизни человека по сравнению с фоновой частотой развития любого вида онкологического заболевания, составляющей 1 к 3. Следует отметить, что существующие оценки канцерогенного риска основаны на пожизненных экспозициях лекарственного препарата. В ходе разработки лекарственного препарата и после его регистрации экспозиция лекарственного препарата меньше, чем пожизненная, поэтому даже в случае повышенного поступления примесей с лекарственным препаратом при его применении в рамках клинической разработки и пострегистрационного обращения сохраняется сопоставимый уровень канцерогенного риска, если такое поступление не превысит допустимый пожизненный уровень. Использование количественного выражения величины канцерогенного риска (1 случай на 100 000) и его пересчет в величины доз, основанные на риске (порог токсикологической угрозы), является условной концепцией, которую не следует рассматривать в качестве реального показателя фактического риска. Тем не менее использование порога токсикологической угрозы позволяет установить уровень безопасной экспозиции для любого мутагенного соединения. При этом следует учитывать, что:
превышение порога токсикологической угрозы необязательно приводит к повышению канцерогенного риска ввиду консервативных допущений, использованных при расчете порога токсикологической угрозы;
наиболее вероятное повышение частоты развития онкологического заболевания гораздо меньше 1 случая на 100 000;
если мутагенное соединение неканцерогенно при испытании на грызунах, прогнозируемое увеличение канцерогенного риска равно нулю;
у пациентов, ранее подвергшихся экспозиции примеси, которая впоследствии идентифицируется как мутагенная, необязательно происходит повышение канцерогенного риска.
В случае применения порога токсикологической угрозы при оценке канцерогенного риска следует запланировать и выполнять дальнейшие действия по контролю канцерогенного риска при применении лекарственного препарата.
14. Если в отношении примеси выявлен потенциальный канцерогенный риск, в целях обеспечения содержания мутагенной примеси на уровне, равном допустимому канцерогенному риску или ниже него, следует разработать надлежащую стратегию контроля, в которой будет учтено понимание процесса управления канцерогенным риском и (или) усиление аналитических методов контроля.
15. В некоторых случаях примесь может являться метаболитом фармацевтической субстанции (действующего вещества), и эту примесь можно квалифицировать с помощью оценки канцерогенного риска, учитывающей мутагенные и канцерогенные свойства образующегося метаболита.
IV. Оценка примесей в зарегистрированных
лекарственных препаратах
16. Настоящее Руководство не применяется к лекарственным препаратам, зарегистрированным в соответствии с правом Евразийского экономического союза (далее - Союз) до даты начала применения настоящего Руководства, за исключением случаев, указанных в настоящем разделе. Если в регистрационные досье лекарственных препаратов, зарегистрированных до даты начала применения настоящего Руководства, вносятся пострегистрационные изменения, перечисленные в настоящем разделе, то они требуют проведения повторной оценки безопасности в отношении мутагенных примесей в соответствии с настоящим разделом.
1. Изменение химических свойств, процесса производства
и контроля качества фармацевтических субстанций
на пострегистрационном этапе
17. Вносимые в регистрационное досье сведения касающиеся проведенных изменений химических свойств, процесса производства и контроля качества фармацевтических субстанций, включают в себя оценку потенциального канцерогенного риска, связанного с мутагенными примесями, появляющимися на всех стадиях начиная с исходного материала в результате изменения:
метода синтеза фармацевтических субстанций;
вида или содержания реагентов;
вида или содержания растворителей;
условий процесса производства.
Следует оценить вероятность образования новых мутагенных примесей или необходимость ужесточения критериев приемлемости имеющихся мутагенных примесей. Повторная оценка примесей, не связанных с такими изменениями не проводится. Например, при внесении изменений, касающихся только части процесса производства, оценку канцерогенного риска мутагенных примесей следует ограничить:
определением вероятности образования новых мутагенных примесей;
оценкой роста содержания имеющихся мутагенных примесей на затронутой изменением стадии производства;
оценкой роста содержания имеющихся мутагенных примесей, образовавшихся на предыдущих стадиях производства.
При подаче в регистрационные досье сведений о таких изменениях, их следует охарактеризовывать в обосновании предлагаемой спецификации, указанном в подразделе 2 раздела IX настоящего Руководства. Не проводится повторная оценка риска появления мутагенных примесей при изменении:
производственной площадки, на которой производится фармацевтическая субстанция;
промежуточных продуктов;
исходных материалов;
поставщика сырья.
18. Если предлагается новый поставщик фармацевтической субстанции, доказательством приемлемого соотношения "польза - риск" в отношении мутагенных примесей и отсутствия необходимости проведения оценки в соответствии с настоящим Руководством является подтверждение того, что фармацевтическая субстанция производится данным поставщиком с использованием того же пути синтеза, что и для других лекарственных препаратов с данной фармацевтической субстанцией, зарегистрированных в рамках Союза. Если это условие не соблюдается, оценка проводится в соответствии с настоящим Руководством.
2. Пострегистрационные изменения сведений о химических
свойствах, процессе производства и контроле качества
лекарственных препаратов
19. Вносимые в регистрационное досье сведения касающиеся проведенных изменений в лекарственном препарате (например, изменение состава, процесса производства, лекарственной формы), включают в себя оценку потенциального канцерогенного риска, связанного с новыми мутагенными продуктами деградации или повышением критериев приемлемости для имеющихся мутагенных продуктов деградации. При необходимости, такие изменения должны содержать обновленную стратегию контроля. При отсутствии изменений фармацевтической субстанции, входящей в состав лекарственного препарата, проведение ее повторной оценки не выполняется. При изменении производственной площадки на которой производится лекарственный препарат не проводится повторная оценка риска, связанного с мутагенными примесями.
3. Изменение области клинического применения
зарегистрированных лекарственных препаратов
или показаний для их применения
20. Изменение области клинического применения зарегистрированных лекарственных препаратов или показаний для их применения, при котором проводится повторная оценка предельного содержания мутагенных примесей, включает в себя:
значительное увеличение суточной дозы лекарственного препарата;
увеличение продолжительности применения (особенно в случае, если мутагенная примесь контролировалась на уровне, который превышал пожизненное допустимое поступление по предыдущему показанию и который уже может не соответствовать более длительному сроку терапии, по новому показанию к применению);
изменение показания к применению при лечении тяжелого или угрожающего жизни заболевания, для которого было обосновано более высокое допустимое поступление, на показание к применению при лечении менее тяжелого заболевания, для которого такое допустимое поступление мутагенных примесей может быть уже неприемлемым.
При изменении показаний для клинического применения зарегистрированных лекарственных препаратов, связанных с новыми путями введения или расширением популяции пациентов (включение беременных женщин и (или) детей), не проводится повторная оценка предельного содержания мутагенных примесей при условии отсутствия повышения суточной дозы или продолжительности терапии.
4. Прочие виды изменений в регистрационном досье
зарегистрированных лекарственных препаратов
21. Настоящее Руководство применяется в отношении зарегистрированных лекарственных препаратов относительно безопасности которых возникают особые опасения. Определение у примеси мутагенного потенциала только на основании характеристики химической структуры считается недостаточным основанием для принятия мер контроля, если такая химическая структура не входит в группу примесей, вызывающую опасения. Вместе с тем особым поводом для опасения являются новые релевантные данные об опасности примеси (класса 1 или 2 согласно таблице 1), полученные после утверждения общей стратегии контроля и спецификаций для регистрации лекарственного препарата. Эти новые релевантные данные об опасности примеси получают в результате научных исследований, проводимых в соответствии с требованиями руководств по токсикологическим исследованиям, утверждаемых Евразийской экономической комиссией, с обеспечением доступа к полным отчетам об этих научных исследованиях и к первичным данным этих научных исследований. Аналогичным образом новая обнаруженная примесь, являющаяся известным мутагеном класса 1 или 2 согласно таблице 1, содержащаяся в зарегистрированном лекарственном препарате, может также быть поводом для опасений. В обоих случаях, если производителю лекарственного препарата становится известна подобная информация, следует провести оценку примесей в соответствии с настоящим Руководством.
V. Оценка примесей в фармацевтической субстанции
и лекарственном препарате до их регистрации
22. Следует провести оценку фактических примесей и примесей с мутагенным потенциалом, которые могут образоваться в ходе синтеза и хранения новой фармацевтической субстанции и при производстве и хранении нового лекарственного препарата. Оценка примесей проводится в 2 этапа:
оценка мутагенного потенциала фактических примесей (то есть фактически присутствующих в фармацевтической субстанции идентифицированных примесей);
оценка потенциальных примесей (то есть определение примесей, которые, с высокой долей вероятности, могут присутствовать в конечной фармацевтической субстанции и определение необходимости дальнейшего изучения их мутагенного потенциала).
1. Оценка примесей, образующихся в ходе синтеза
23. К фактическим примесям относятся примеси, содержание которых в фармацевтической субстанции превышает пороги идентификации, предусмотренные правилами по изучению примесей в лекарственных средствах и установлению требований к ним в спецификациях, определяемыми Комиссией (далее - пороги идентификации). Идентификация фактических примесей выполняется, если их содержание в фармацевтической субстанции превышает пороги идентификации. К идентифицированным примесям (в рамках изучения мутагенности) могут быть отнесены и некоторые примеси, содержание которых в фармацевтической субстанции ниже порога идентификации.
24. К потенциальным примесям в фармацевтической субстанции относятся исходные материалы, реагенты и образующиеся из исходного материала промежуточные продукты в процессе синтеза фармацевтической субстанции. Следует оценить риск переноса в фармацевтическую субстанцию идентифицированных примесей, содержащихся в исходных материалах и промежуточных продуктах, и примесей, обоснованно считающихся побочными продуктами для использованного пути синтеза фармацевтической субстанции из исходного материала. Поскольку для некоторых примесей (например, образующихся на ранних стадиях многостадийного синтеза) риск переноса в фармацевтическую субстанцию может быть ничтожным, следует представить обоснование вероятности их переноса на основе оценки такого риска выполненной для каждой точки (стадии) пути синтеза и определить ту точку (стадию) пути синтеза после которой следует выполнять оценку мутагенного потенциала этого вида примесей.
25. Для исходных материалов, вводимых на поздних стадиях синтеза фармацевтической субстанции (если путь синтеза исходного материала известен), следует провести оценку завершающих стадий процесса синтеза исходного материала на наличие потенциальных мутагенных примесей.
26. Следует провести оценку фактических идентифицированных примесей с известной химической структурой и потенциальных примесей, описанных в настоящем подразделе, на предмет мутагенного потенциала в соответствии с разделом VI настоящего Руководства.
2. Оценка продуктов деградации
27. К фактическим продуктам деградации лекарственного препарата относятся продукты деградации, которые при хранении лекарственного препарата в предлагаемых условиях долгосрочного хранения в первичной, и во вторичной упаковках превышают порог идентификации, а также примеси, которые образуются при производстве лекарственного препарата.
28. Идентификацию фактических продуктов деградации следует проводить при превышении ими порогов идентификации. Допускается идентификация в качестве фактических продуктов деградации некоторых продуктов деградации, содержание которых в лекарственном препарате ниже порога идентификации. Потенциальными продуктами деградации фармацевтической субстанции и лекарственного препарата являются продукты деградации, образование которых обоснованно ожидается в условиях долгосрочного хранения. К потенциальным продуктам деградации относятся продукты деградации, образование которых превышает порог идентификации в исследованиях ускоренного хранения (например, при температуре 40 +/- 2 °C и относительной влажности 75 +/- 5% в течение 6 месяцев) и в подтверждающих исследованиях фотостабильности, описанных в Требованиях к исследованию стабильности лекарственных препаратов и фармацевтических субстанций, утвержденных Решением Коллегии Евразийской экономической комиссии от 10 мая 2018 г. N 69, и наличие которых в фармацевтической субстанции или лекарственном препарате в условиях долгосрочного хранения в первичной упаковке еще не подтверждено.
29. При принятии решения по выбору потенциальных продуктов деградации для оценки их мутагенности используются сведения о релевантных путях деградации (например, на основе принципов химической деградации, релевантных исследований в стрессовых условиях и исследований стабильности при разработке лекарственного препарата).
30. Фактические и потенциальные продукты деградации с известной химической структурой, которые с высокой долей вероятности будут содержаться в конечной фармацевтической субстанции или готовом лекарственном препарате, следует оценить на предмет их мутагенного потенциала в соответствии с разделом VI настоящего Руководства.
3. Вопросы клинической разработки лекарственного препарата
31. Оценка примесей и потенциальных продуктов деградации, описанных в подразделах 1 и 2 настоящего раздела, применяется для лекарственных препаратов, находящихся на этапе клинической разработки, несмотря на ограниченность сведений об их химической структуре и потенциальной мутагенности. Например, в ходе клинической разработки могут отсутствовать результаты долгосрочных исследований стабильности и фотостабильности, поэтому сведения о потенциальных продуктах деградации могут быть недостаточными. К лекарственным препаратам, находящимся на этапе клинической разработки, не применяются дополнительные пороги идентификации, следовательно, на этих этапах будет идентифицировано меньшее количество примесей по сравнению с лекарственными препаратами, прошедшими регистрацию.
VI. Элементы оценки опасности примесей
32. Оценка опасности примесей предполагает первичный анализ фактических и потенциальных примесей путем проведения поиска по базам данных и научным источникам информации о мутагенности и канцерогенности примесей в бактериальных тест-системах, что позволяет классифицировать примеси, относя их к 1, 2 или 5 классу согласно таблице 1. Если такие классификационные данные отсутствуют, следует провести оценку количественной структурно-функциональной зависимости (QSAR), которая позволяет прогнозировать наличие мутагенности в бактериальных тест-системах. Это позволит классифицировать примеси, отнеся их к 3, 4 или 5 классу согласно таблице 1.
Таблица 1
Классификация примесей по мутагенному и канцерогенному
потенциалу и соответствующие меры контроля
Класс
Определение
Меры контроля
1
Известные мутагенные канцерогены
контроль на уровне, который равен или ниже специфичному для соединения допустимому пределу содержания
2
Известные мутагены с неизвестным канцерогенным риском (наличие мутагенности в бактериальных тест-системах <*>, отсутствие данных о канцерогенности у грызунов)
контроль на уровне, который равен допустимому или ниже допустимого предела содержания (данный уровень соответствует порогу токсикологической угрозы)
3
Структурный признак, не связанный со структурой фармацевтической субстанции. Отсутствие данных по мутагенности
контроль на уровне, который равен допустимому или ниже допустимого предела (данный уровень соответствует порогу токсикологической угрозы)
либо
проведение теста на мутагенность в бактериальных тест-системах, по результатам которого вещество относят к классу 5 (если оно не мутагенно) или классу 2 (если оно мутагенно)
4
Структурный признак, тот же признак у фармацевтической субстанции или родственных для фармацевтической субстанции соединений (например, промежуточных производственных продуктов), которые подверглись испытанию и оказались немутагенными
рассматривать как немутагенную примесь
5
Отсутствие структурных признаков или структурный признак с данными, подтверждающими отсутствие мутагенности или канцерогенности
рассматривать как немутагенную примесь
--------------------------------
<*> Или наличие других релевантных данных о мутагенности, указывающих на генные мутации, связанные с ДНК-реактивностью (например, положительные результаты генных мутаций исследований in vivo).
33. Следует провести токсикологическую оценку расчетным методом на основе анализа количественной структурно-функциональной зависимости (QSAR), прогнозирующим результат теста бактериальной мутагенности. Следует применить 2 взаимодополняющие методологии анализа количественной структурно-функциональной зависимости (QSAR). Первая методология должна основываться на экспертных оценках, вторая - на статистических данных. Компьютеризированные системы, моделирующие количественные структурно-функциональные зависимости (QSAR), в которых применяются такие методы прогнозирования, должны быть валидированы на основании общих принципов, предусмотренных приложением N 4 к Правилам надлежащей лабораторной практики Евразийского экономического союза в сфере обращения лекарственных средств, утвержденным Решением Совета Евразийской экономической комиссии от 3 ноября 2016 г. N 81 (далее - Правила надлежащей лабораторной практики).
34. Отсутствие структурных признаков мутагенности примеси при применении 2 взаимодополняющих методов оценки количественной структурно-функциональной зависимости (основанных на экспертных оценках и статистических данных) является достаточным основанием для вывода о том, что примесь не вызывает мутагенных опасений, и проведение дальнейших исследований не требуется (класс 5 в таблице 1).
35. В целях получения дополнительных данных, подтверждающих правильность любых положительных, отрицательных, противоречивых или неоднозначных прогнозов и составления научной основы окончательного заключения, результат любого компьютерного анализа следует подвергать дополнительной оценке, используя научные знания (при необходимости).
36. В целях последующего наблюдения за значимым структурным признаком (класс 3 в таблице 1) возможно принятие надлежащих мер контроля или проведение теста на мутагенность бактерий только с воздействием примеси.
37. В целях оценки мутагенного потенциала примесей можно провести 1 тест на мутагенность бактерий, используя подходящий детальный протокол исследований. Тесты следует проводить в соответствии с Правилами надлежащей лабораторной практики, однако допускается при проведении тестов использование данных для обоснования проведения клинических исследований и регистрации лекарственных препаратов и в случае отклонения от требований Правил надлежащей лабораторной практики. Такие отклонения следует описать в отчете об исследовании (например, пробоподготовка или анализ исследуемого препарата могут не соответствовать требованиям Правил надлежащей лабораторной практики). В некоторых случаях выбор штаммов бактерий для тестов допускается ограничить только штаммами, подтвердившими чувствительность к идентифицированному признаку мутагенности. В случае если примеси невозможно выделить или синтезировать или количество примеси ограничено, максимальные испытуемые концентрации таких примесей (которые рекомендуется достигать при оценке мутагенности) могут быть не достигнуты в ходе тестов. В этом случае в целях проведения тестов при высоких концентрациях примеси тест на бактериальную мутагенность можно провести при наличии соответствующего обоснования, используя уменьшенный формат тестов при условии доказательства его высокого соответствия методике исследований, приведенной в документах Организации экономического сотрудничества и развития или Международной конференции по гармонизации технических требований к регистрации лекарственных препаратов для человека.
38. Отрицательный результат выполненного надлежащим образом теста на мутагенность у бактерий позволяет отклонить любое опасение в отношении наличия мутагенной активности, связанной с химической структурой примеси, в связи с чем дальнейшая оценка генотоксичности такой примеси не рекомендуется. Такие примеси следует рассматривать как немутагенные (класс 5 в таблице 1).
39. Подход к оценке способности потенциальных примесей вызывать точечные мутации обеспечивает контроль таких мутаций на безопасном уровне, поэтому если содержание примеси ниже или выше порога квалификации, предусмотренного правилами по изучению примесей в лекарственных средствах и установлению требований к ним в спецификациях, дополнительная квалификация ее мутагенного потенциала не требуется. Такой подход предполагает начальное использование методик оценки количественной структурно-функциональной зависимости для прогнозирования мутагенности в бактериальных тест-системах. Если количество примеси превышает суточную дозу, равную 1 мг, при длительном применении, можно предусмотреть оценку генотоксического потенциала в соответствии с правилами по изучению примесей в лекарственных средствах и установлению требований к ним в спецификациях. Если количество примеси в суточной дозе меньше 1 мг, дальнейшее изучение генотоксичности не требуется, независимо от величины установленных порогов квалификации.
Положительный результат теста на мутагенность у бактерий потребует дальнейшей оценки опасности и (или) принятия мер контроля (класс 2 в таблице 1). Например, если содержание примеси невозможно контролировать на приемлемом уровне, то в целях определения применимости результатов теста на мутагенность у бактерий для исследований in vivo примесь рекомендуется испытать в тесте на генные мутации in vivo. Выбор других тестов на генотоксичность в исследованиях in vivo следует научно обосновать, на основе сведений о механизме действия примеси и ожидаемой ее экспозиции в тканях-мишенях.
Таблица 2
Выбор исследований in vivo для проверки данных
о мутагенности полученных в испытаниях in vitro
(положительная мутагенность в бактериальных тест-системах)
Исследования in vivo
Критерии, обосновывающие выбор теста, соответствующего своему целевому назначению
Тесты на трансгенные мутации
при любых положительных результатах оценки мутагенности в бактериальных тест-системах. Следует обосновать выбор ткани (органа) для испытаний
Тест Pig-a (кровь)
для мутагенов прямого действия (положительная мутагенность в бактериальных тест-системах при отсутствии результатов оценки мутагенности, выполненной на фракции микросом печени S9) <*>
Микроядерный тест (кровь или костный мозг)
для мутагенов прямого действия (положительная мутагенность в бактериальных тест-системах при отсутствии результатов оценки мутагенности, выполненной на фракции микросом печени S9) и кластогенных соединений <*>
Тест внепланового синтеза ДНК (ВСД) в печени крысы
исключительно при положительных результатах оценки мутагенности, выполненной на фракции микросом печени S9
установлено образование мутагенного печеночного метаболита в организме подопытных видов животных в результате воздействия на них исходных химических соединений
Тест ДНК-комет
требуется обоснование использования данного теста:
выбор теста обусловлен способом действия мутагенного соединения;
тест является специфичным для данного химического класса соединений;
под воздействием соединения образуются неустойчивые  участки или одноцепочечные разрывы, предшествующие повреждению ДНК, которое может приводить к мутациям.
участки или одноцепочечные разрывы, предшествующие повреждению ДНК, которое может приводить к мутациям.
следует обосновать выбор ткани (органа) для испытаний
Прочие
при представлении научно обоснованных данных о применимости теста для оценки мутагенности соединения
--------------------------------
<*> Для мутагенов непрямого действия (требующих метаболической активации) необходимо показать достаточность экспозиции образующихся из них метаболитов.
Исследования in vivo следует планировать, принимая во внимание существующие акты по генотоксичности Евразийской экономической комиссии. Результаты надлежащим образом выполненного теста in vivo, могут служить основой для установления примесей в составе какого-либо соединения специфичных пределов содержания.
40. Примесь, имеющая общее с фармацевтической субстанцией или родственными соединениями строение (например, одинаковую структурную функциональную группу в положении заместителя в родоначальной структуре и идентичное химическое окружение), можно считать немутагенной (класс 4 в таблице 1) при отрицательных результатах испытания этой примеси в тесте на мутагенность у бактерий.
VII. Установление характеристик рисков
41. По результатам оценки опасности каждая примесь будет отнесена к одному из пяти классов согласно таблице 1. В настоящем разделе описаны принципы установления характеристик рисков, используемые для расчета допустимого поступления примесей классов 1, 2 и 3.
1. Определение допустимого поступления, основанное
на пороге токсикологической угрозы
42. Риск допустимого поступления мутагенной примеси, основанный на пороге токсикологической угрозы, равном 1,5 мкг на человека в сутки, считается ничтожным (теоретическое повышение канцерогенного риска менее 1 случая на 100 000 при пожизненной экспозиции), что позволяет по умолчанию осуществлять расчет допустимого предела для контроля содержания примесей в составе большинства лекарственных препаратов. Этот подход обычно используется в отношении мутагенных примесей, содержащихся в лекарственных препаратах для длительного применения (более 10 лет), при отсутствии данных о канцерогенности (классы 2 и 3 согласно таблице 1).
2. Определение допустимого поступления, основанное на оценке
рисков, специфичных для конкретного химического соединения
Оценка мутагенных примесей с установленной
канцерогенностью (класс 1 в таблице 1)
43. При наличии достаточных данных о канцерогенности для расчета допустимого поступления взамен применения подхода, основанного на пороге токсикологической угрозы, следует прибегнуть к оценке рисков, специфичных для конкретного химического соединения. Специфичное для химического соединения допустимое поступление известного мутагенного канцерогена можно по умолчанию рассчитать, основываясь на канцерогенной активности и линейной экстраполяции.
44. Путем линейной экстраполяции можно рассчитать специфичное для химического соединения допустимое поступление в организм человека, основанное на определении величины ТД50, установленной при оценке канцерогенной активности этого соединения у грызунов. Линейная экстраполяция до вероятности 1 случай на 100 000 (то есть использованный допустимый уровень пожизненного риска) достигается простым делением значения ТД50 на 50 000. Эта процедура аналогична вычислению порога токсикологической угрозы.
Пример расчета для этиленоксида.
Значение ТД50 для этиленоксида согласно базе данных канцерогенной активности составляет 21,3 мг/кг массы тела в сутки (крысы) и 63,7 мг/кг массы тела в сутки (мыши). Для расчета допустимого поступления этиленоксида в организм человека использовано меньшее (то есть более консервативное) значение, установленное для крыс.
Для расчета дозы, вызывающей образование опухоли у 1 животного из 100 000, необходимо поделить значение ТД50 на 50 000:
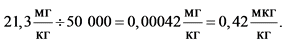
Для расчета общей суточной дозы для человека следует пересчитать на массу тела человека:
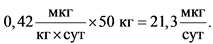
Следовательно, ежедневное пожизненное поступление 21,3 мкг этиленоксида будет соответствовать теоретическому канцерогенному риску, равному 10-5 (1 случай на 100 000) и будет допустимым поступлением при его наличии в виде примеси в составе лекарственного вещества.
В качестве альтернативы допускается применение других устоявшихся методик оценки рисков (например, методики оценки риска для проведения расчета приемлемого поступления мутагенных соединений, используемые международными организациями по контролю безопасности химических соединений, или использование установленных этими организациями значений приемлемого поступления мутагенных соединений).
Опубликованные данные по установленным пределам воздействия для оценки канцерогенного риска могут быть использованы в качестве альтернативы применению наиболее консервативного значения ТД50, полученного на основании исследований канцерогенности на грызунах, независимо от значимости уровня таких пределов воздействия для человека. При использовании результатов токсикологической оценки на животных в качестве отправной точки для линейной экстраполяции канцерогенного риска у человека, в целях первичного выявления результатов с наибольшей значимостью (в зависимости от вида животного, органа-мишени и т.д.) следует провести детальную экспертную токсикологическую оценку имеющихся данных о канцерогенности. В целях улучшения результатов прогнозирования, исходя из формы кривой зависимости "доза - эффект", в качестве исходного численного показателя канцерогенной активности примеси вместо значений ТД50 допускается использовать контрольную дозу (в качестве контрольной дозы может использоваться нижний 10% доверительный уровень (BMDL10), соответствующий наименьшей дозе, которая с вероятностью 95% приведет к развитию онкологического заболевания у крыс с частотой не более чем 10%). В таком случае линейная экстраполяция этой дозы на дозу у человека, вызывающую вероятность развития онкологического заболевания с частотой 1 случай на 100 000 (то есть, соответствующей допустимому пожизненному уровню риска), достигается простым делением значения нижнего доверительного уровня (BMDL10) на 10 000.
Специфичное для конкретного химического соединения допустимое поступление мутагенных примесей допускается рассчитывать на основании значений токсикологических порогов для таких соединений, указанных в документах принятых международными организациями, занимающимися вопросами безопасности химических соединений (технические доклады Всемирной организации здравоохранения, Программа оценки канцерогенного риска в рамках Международной программы химической безопасности (МПХБ) и т.д.), при этом следует использовать пожизненный уровень риска, соответствующий величине 10-5 (1 случай на 100 000). Используемый при таком методе оценки токсикологический порог должен основываться на последних научных данных и (или) методологии оценки токсикологических порогов.
45. В отношении примесей сходной химической структуры допускается проводить определение допустимого поступления одного вида примесей на основании токсикологических пределов поступления установленных для другого химически родственного вида канцерогенных примесей (специфичное для определенного класса допустимое поступление) при условии, что имеются научное обоснование химической схожести примесей и данные, обосновывающие возможность применения такого подхода.
46. В отношении мутагенных примесей (при отсутствии данных об их канцерогенности) допускается применять методику расчета класс-специфичного поступления, описанную в пункте 45 настоящего Руководства, при условии, что химическое строение мутагенных примесей идентично химическому строению какого-либо из известных классов канцерогенных соединений. Например, известную канцерогенную активность производных полифункциональных алкилхлоридов можно использовать для обоснования расчета безопасного допустимого поступления примесей производных монофункциональных алкилхлоридов (примесей группы алкилхлоридов), образующихся в процессе синтеза лекарственных веществ. Монофункциональные алкилхлоридные соединения являются слабыми канцерогенами по сравнению с производными полифункциональных алкилхлоридов, для которых величина ТД50 находится в диапазоне от 36 до 1 810 мг/кг/сут. Данная величина для полифункциональных алкилхлоридов установлена на основе данных, полученных при изучении 15 тест-объектов из соединений данной группы, за исключением эпихлорогидрина, относящегося к бифункциональным алкилхлоридам с 2 полностью различающимися функциональными группами). Таким образом, значение ТД50 равное 36 мг/кг/сут допустимо использовать в качестве очень консервативной оценки класс-специфичной референтной точки канцерогенной активности для расчета допустимого поступления монофункциональных алкилхлоридов. Этот уровень канцерогенной активности, по меньшей мере в 10 раз ниже, чем значение верхней границы диапазона ТД50 равной 1,25 мкг/кг/сут, и не превышает базовый пожизненный порог токсикологической угрозы (1,5 мкг/сут). Следовательно, это значение позволяет обосновать пожизненное и менее чем пожизненное ежедневное поступление монофункциональных алкилхлоридов в количестве которое в 10 раз меньше, чем базовый пожизненный порог токсикологической угрозы.
Оценка мутагенных примесей при наличии данных
о практическом пороге их негативного воздействия
47. При оценке мутагенных примесей допускается использовать подход, основанный на установлении дозы, не приводящей к развитию наблюдаемых эффектов (NOEL), или на расчете разрешенной дневной экспозиции примеси с использованием коэффициентов неопределенности в случае наличия у химического соединения:
а) механизмов, приводящих к нелинейной зависимости "доза - эффект";
б) зависимости порога негативного воздействия химического соединения от его влияния на молекулы-мишени, не являющиеся ДНК;
в) модуляции токсического действия мутагенных примесей за счет их быстрой инактивации до контакта с ДНК или за счет эффективной репарации произошедшего повреждения.
48. Допустимое поступление мутагенных примесей, рассчитанное на основании специфичной для химического соединения оценки рисков, можно:
скорректировать для более короткой продолжительности применения лекарственного препарата с учетом показателей, приведенных в подразделе 3 настоящего раздела;
ограничить на уровне в 0,5% и менее.
При этом из двух вышеперечисленных подходов должен быть выбран тот, который позволяет получить более низкое значение.
Например, если специфичное для химического соединения допустимое суточное поступление мутагенной примеси составляет 15 мкг/сут при пожизненной экспозиции, то менее чем пожизненные пределы содержания (приведенные в таблице 3) можно увеличить до суточного поступления мутагенной примеси, равного 100 мкг (при условии, что продолжительность терапии составит 1 - 10 лет), 200 мкг (при условии, что продолжительность терапии составит 1 - 12 месяцев) или 1 200 мкг (при условии, что продолжительность терапии не превысит 1 месяца). Однако для лекарственного препарата с максимальной суточной дозой 100 мг допустимое суточное поступление мутагенной примеси при продолжительности терапии менее 1 месяца будет ограничено величиной 0,5% (0,5 мг или 500 мкг, а не 1 200 мкг).
3. Определение допустимого поступления мутагенной
примеси при менее чем пожизненной экспозиции вещества
49. Стандартная оценка рисков известных канцерогенов показывает, что канцерогенный риск возрастает в зависимости от накопленной дозы мутагенной примеси. Таким образом, риск непрерывного пожизненного поступления небольшой дозы мутагенной примеси эквивалентен канцерогенному риску, обусловленному идентичной накопленной экспозицией мутагенной примеси, полученной за меньший период времени.
50. Основанное на пороге токсикологической угрозы допустимое поступление мутагенной примеси равное 1,5 мкг/сут, считается безопасным при ее пожизненной ежедневной экспозиции. Чтобы обеспечить поступление мутагенной примеси в лекарственном препарате на уровне меньше чем пожизненная экспозиция используется подход при котором приемлемое накопительное пожизненное поступление мутагенной примеси равными частями распределяется на общее число дней, которое меньше чем пожизненная экспозиция. Такой подход позволяет даже в условиях более высокого суточного поступления мутагенных примесей, по сравнению с величиной их пожизненного поступления, сохранить риск мутагенности на сопоставимом уровне при ежедневном приеме лекарственного препарата и при периодическом (неежедневном) приеме лекарственного препарата. Для расчета пожизненного поступления мутагенной примеси в течение 70 лет ее накопленное поступление для уровня 1,5 мкг/сут составит: 1,5 мкг/сут x 25 550 дней = 38,3 мг. В таблице 3 приведено допустимое поступление примеси лекарственного препарата при его пожизненной экспозиции и при экспозиции меньшей чем пожизненная, как в рамках клинической разработки лекарственного препарата, так и после его регистрации. При периодическом применении лекарственного препарата допустимое суточное поступление мутагенной примеси следует определять на основании общего количества дней применения лекарственного препарата, а не на основании  интервала в котором осуществлялось применение лекарственного препарата, и это количество дней применения лекарственного препарата должно соотноситься с соответствующей категорией продолжительности терапии, указанной в таблице 3. Например, допустимое поступление примеси от лекарственного препарата, применяемого 1 раз в неделю в течение 2 лет (то есть 104 дня применения), будет составлять 20 мкг/сут.
интервала в котором осуществлялось применение лекарственного препарата, и это количество дней применения лекарственного препарата должно соотноситься с соответствующей категорией продолжительности терапии, указанной в таблице 3. Например, допустимое поступление примеси от лекарственного препарата, применяемого 1 раз в неделю в течение 2 лет (то есть 104 дня применения), будет составлять 20 мкг/сут.
Таблица 3
Допустимое поступление в организм человека
для каждой отдельной мутагенной примеси
в составе лекарственного препарата
Продолжительность терапии
<= 1 месяца
> 1 - 12
месяцев
> 1 - 10
лет
> 10 лет или пожизненно
Суточное поступление (мкг/сут)
120
20
10
1,5
Оценка примесей в рамках клинической
разработки лекарственного препарата
51. В рамках клинической разработки лекарственного препарата вплоть до завершения клинических исследований III фазы уровень допустимого поступления каждой отдельной мутагенной примеси для различных сроков терапии установленный на основе концепции поступления мутагенной примеси в количестве меньшем чем пожизненная экспозиция приведен в таблице 3. Эти скорректированные значения допустимого поступления мутагенной примеси обеспечивают:
уровень риска, равный 1 случаю на 1 000 000 для ранней стадии клинической разработки лекарственного препарата, когда его польза еще не установлена;
уровень риска, равный 1 случаю на 100 000 для более поздних стадий клинической разработки.
52. До установления порога поступления мутагенных примесей меньшего, чем их пожизненное допустимое поступление следует определить пределы порогов токсикологической угрозы для каждой стадии клинической разработки лекарственных препаратов. Расчет порога поступления мутагенных примесей, который меньше чем пожизненное допустимое поступление (ДП) основывается на применении правила Хабера, согласно которому произведение концентрации (C) поступающего в организм соединения и времени воздействия (T) этого соединения - есть величина постоянная (k):
C x T = k.
Исходя из этого канцерогенный эффект любого соединения зависит как от его дозы (величины поступления), так и длительности экспозиции.
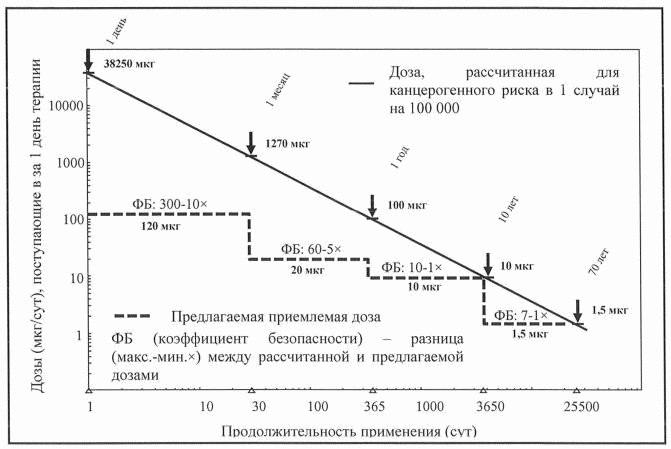
Рисунок 1. Зависимость рассчитанного суточного поступления
мутагенной примеси от продолжительности применения
лекарственного препарата соответствующее теоретическому
канцерогенному риску, равному 1 случаю на 100 000
Суточное поступление мутагенной примеси является функцией отражающей зависимость продолжительности терапии от допустимого поступления этой примеси. Сплошная линия на рисунке 1 представляет линейный вид такой зависимости от количества дней терапии для поступления мутагенной примеси, соответствующей канцерогенному риску, равному 1 случаю на 100 000 (или риску в 10-5). Расчет основан на пороге токсикологической угрозы, применяемом в настоящем Руководстве для пожизненной терапии, то есть уровне поступления в 1,5 мкг/сут по следующей формуле:
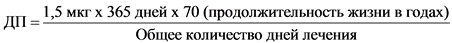
Тогда расчетное суточное поступление будет составлять:
1,5 мкг при продолжении поступления лекарственного препарата в течение всей жизни (70 лет);
10,5 мкг - при продолжении поступления лекарственного препарата в течение 10 лет;
105 мкг - при продолжении поступления лекарственного препарата в течение 1 года;
1 277,5 мкг - при продолжении поступления лекарственного препарата в течение 1 месяца;
38 325 мкг (примерно 38,3 мг) - при однократном применении лекарственного препарата.
Во всех вышеперечисленных случаях будет обеспечено одинаковое кумулятивное поступление мутагенной примеси и, следовательно, теоретически одинаковый канцерогенный риск (1 случай на 100 000).
Пунктирная кривая на рисунке 1 представляет фактическое суточное поступление, скорректированное для экспозиции, которая меньше чем пожизненная экспозиция в соответствии с рекомендациями раздела VII настоящего Руководства для лекарственных препаратов, находящихся в процессе клинической разработки, и зарегистрированных лекарственных препаратов. Эти предлагаемые уровни в целом значительно ниже расчетных значений, что позволяет получить коэффициенты безопасности, увеличивающиеся по мере сокращения продолжительности терапии.
Предлагаемое допустимое суточное поступление мутагенной примеси также соответствует канцерогенному риску, равному 1 случай на 1 000 000 (или риску в 10-6), если продолжительность терапии не превышает 6 месяцев и, если такое поступление рассчитано для ранних стадий клинических исследований у добровольцев или пациентов, когда польза лекарственного препарата еще не установлена. В этом случае коэффициенты безопасности, показанные в на рисунке 1, уменьшаются в 10 раз.
53. В клинических исследованиях I фазы с продолжительностью применения лекарственного препарата до 14 дней можно применить альтернативный подход по отношению к консервативному использованию скорректированного допустимого поступления любой мутагенной примеси. При таком подходе следует обеспечить контроль соответствия допустимым пределам, описанным в разделе VIII настоящего Руководства, только содержания тех примесей, которые являются известными мутагенными канцерогенами (класс 1 в таблице 1) и известными мутагенами с неизвестным канцерогенным потенциалом (класс 2 в таблице 1), а также примесей из класса химических веществ, входящего в группу, вызывающую опасения.
Все другие примеси рассматриваются как немутагенные примеси. К ним относятся примеси, которые содержат структурные признаки токсичности (класс 3 в таблице 1) и сами по себе не требуют оценки в рамках ограниченной продолжительности исследований I фазы.
Оценка примесей в зарегистрированных
лекарственных препаратах
54. Сроки продолжительности терапии с допустимым поступлением примесей, указанные в таблице 3 для зарегистрированных лекарственных препаратов, предназначены для применения к ожидаемой продолжительности экспозиции у большинства пациентов. Предлагаемое поступление примесей в сочетании с различными сценариями такого поступления описаны в таблице 4.
Таблица 4
Примеры сценариев с различной продолжительностью
клинического применения лекарственного препарата
для установления уровня допустимого поступления примесей
Сценарий <*>
Допустимое поступление
(мкг/сут)
Продолжительность терапии <= 1 месяца: например, экстренно применяемые средства (антидоты, анестезия, купирование острого ишемического инфаркта мозга), сенильный кератоз, педикулез
120
Продолжительность терапии > 1 - 12 месяцев: например, противомикробная терапия (вирусного гепатита C) с максимальной продолжительностью до 12 месяцев, препараты для парентерального питания; препараты для профилактики гриппа (~ 5 месяцев), препараты для лечения пептической язвы желудка, вспомогательные репродуктивные технологии (ВРТ), препараты, назначаемые при преждевременных родах, при эклампсии беременных, предоперационная терапия (при гистерэктомии), препараты для лечения переломов (краткосрочное применение, но с длительными периодами полувыведения)
20
Продолжительность терапии > 1 - 10 лет: - например, стадия заболевания с короткой ожидаемой продолжительностью жизни (тяжелые стадии болезни Альцгеймера), негенотоксичная противоопухолевая терапия, применяемая у пациентов с ожидаемой длительной продолжительностью жизни (рак молочной железы, хронический миелолейкоз), препараты, специально предназначенные для менее чем 10-летнего применения, препараты, применяемые периодически для лечения острых рецидивирующих заболеваний <**> (хронический герпес, приступы подагры, лечение лекарственной зависимости (в том никотиновой зависимости), макулярная дегенерация, ВИЧ-инфекция <***>
10
Продолжительность терапии > 10 лет или пожизненная терапия: например, показания для постоянного применения с высокой вероятностью пожизненного применения в широком возрастном диапазоне (артериальная гипертензия, дислипидемия, бронхиальная астма, болезнь Альцгеймера (за исключением тяжелых стадий болезни Альцгеймера), гормональная терапия (например, гормоны роста, гормоны щитовидной железы, паратиреоидный гормон), липодистрофия, шизофрения, депрессия, псориаз, атопический дерматит, хроническая обструктивная болезнь легких (ХОБЛ), муковисцидоз (кистозный фиброз), сезонный и круглогодичный аллергический ринит
1,5
--------------------------------
<*> В таблице приведены типовые примеры. Каждый пример следует анализировать отдельно. Например, доза 10 мкг/сут может быть применима в случаях ограниченной ожидаемой продолжительности жизни пациента (например, в случае тяжелой стадии болезни Альцгеймера), даже если применение лекарственного препарата может превысить 10 лет.
<**> Периодическое применение лекарственного препарата в течение более 10 лет, но на основании рассчитанной накопленной дозы примеси ее поступление, попадает в категорию "> 1 - 10 лет".
<***> ВИЧ-инфекция считается хроническим заболеванием, но после 5 - 10 лет применения к лекарственным препаратам формируется резистентность, и следует переходить на другие препараты для лечения ВИЧ-инфекции, поэтому продолжительность применения лекарственных препаратов в этой группе пациентов не попадает в категорию "более 10 лет или пожизненная терапия".
55. В некоторых случаях допускается продлевать применение зарегистрированных лекарственных препаратов на протяжении большего срока, чем предельный срок продолжительности терапии (например, при расчете допустимого поступления примеси равного 10 мкг/сут исходя из продолжительности терапии лекарственным препаратом 1 - 10 лет следует учесть вероятного того, возможно ли продлить терапию свыше 10 лет (например, до 15 лет)). Такое продление терапии должно гарантировать совсем незначительный прирост риска мутагенности по сравнению с общим рассчитанным риском для большинства пациентов, получающих терапию в течение 10 лет (в приведенном примере риск мутагенности увеличивается до 1,5 случаев на 100 000).
Подходы к применению настоящего Руководства в отношении различных видов зарегистрированных лекарственных препаратов приведены в приложении N 1 к настоящему Руководству.
4. Оценка допустимого поступления нескольких
мутагенных примесей
56. К каждой отдельно взятой примеси следует применять основанное на пороге токсикологической угрозы допустимое поступление. При наличии двух примесей класса 2 или класса 3 применяются индивидуальные пределы. Если в спецификацию на фармацевтическую субстанцию (лекарственный препарат) включены 3 и более примесей класса 2 или класса 3, суммарное поступление мутагенных примесей следует ограничить величинами, представленными в таблице 5 для лекарственных препаратов, находящихся на этапе клинической разработки и зарегистрированных лекарственных препаратов.
57. Поступление примеси каждого действующего вещества, входящего в состав комбинированных лекарственных препаратов подлежит раздельному нормированию.
Таблица 5
Допустимое суточное поступление в организм человека
для нескольких примесей в составе лекарственного препарата
Продолжительность терапии
<= 1 месяца
> 1 - 12
месяцев
> 1 - 10
лет
> 10 лет или пожизненно
Суточное поступление (мкг/сут)
120
60
30
5
58. В расчет суммарного суточного поступления включены только примеси классов 2 и 3 из всех примесей указанных в спецификации на фармацевтическую субстанцию. Однако примеси со специфичными для соединения пределами допустимого поступления или пределами, установленными на основании класса (класс 1) не следует включать в суммарные пределы содержания примесей класса 2 и класса 3. Кроме того, продукты деградации, образующиеся в лекарственном препарате, подлежат отдельному контролю и суммарный предел к ним не применяется.
5. Исключения из консервативного подхода к оценке
примесей (гибкий подход при оценке примесей)
59. Повышенное допустимое поступление какой-либо примеси может быть обосновано, если при поступлении из других источников (например, с пищей или в результате эндогенного метаболизма, как у формальдегида) экспозиция этой примеси у человека будет больше.
60. Индивидуальные исключения из консервативного подхода к оценке надлежащего допустимого поступления примесей допускается обосновывать в случаях, если:
лекарственный препарат применяется при лечении тяжелого заболевания;
у пациентов, принимающих лекарственный препарат, ожидается короткая продолжительность жизни;
для хронического заболевания характерен поздний дебют или при его лечении ограничены терапевтические возможности.
61. Соединения некоторых структурных классов мутагенов могут проявлять чрезвычайно высокую канцерогенную активность (группа, вызывающая опасения), например, афлатоксиноподобные соединения, N-нитрозосоединения и алкил-азоксисоединения. В случае если эти соединения обнаруживаются в виде примесей в лекарственных препаратах, допустимое поступление для этих высокоактивных канцерогенов должно быть существенно ниже, чем допустимое поступление, определенное настоящим Руководством для обычных мутагенных примесей. Несмотря на то, что методы, описанные в настоящем Руководстве, применяются в целях обоснования допустимого поступления примесей в составе лекарственных препаратов, находящихся на этапе фармацевтической разработки, и зарегистрированных лекарственных препаратов, производитель вправе разработать индивидуальный подход к нормированию поступления примесей (например, с использованием данных о канцерогенности близкородственных структур (при наличии таких данных)).
62. Описанные в настоящем Руководстве подходы к оценке рисков применимы ко всем путям введения лекарственных препаратов и в целом не требуют коррекции уровня допустимого поступления примесей в зависимости от пути введения лекарственного препарата. Исключения могут касаться ситуаций, когда воздействие примесей имеет специфические особенности, обусловленные путем введения лекарственного препарата, которые следует оценивать в индивидуальном порядке. Описанные в настоящем Руководстве подходы к оценке рисков также применимы для любых популяций пациентов, подвергшихся воздействию примесей, поскольку для нормирования уровня допустимого поступления примесей использованы консервативные методы расчетов.
VIII. Контроль уровня допустимого поступления примесей
63. Стратегия контроля уровня допустимого поступления примесей включает в себя в том числе следующее:
а) контроль качества материалов (включая сырье, исходные материалы, промежуточные продукты, реагенты, растворители, материалы первичной упаковки);
б) условия эксплуатации производственной площадки и оборудования;
в) контрольные меры, предусмотренные при планировании процесса производства лекарственного препарата;
г) внутрипроизводственные методы контроля (включая внутрипроизводственные испытания и контроль параметров процесса);
д) контроль качества фармацевтической субстанции и лекарственного препарата (например, выпускающие испытания).
64. Если примесь отнесена к классам 1, 2 или 3 в соответствии с таблицей 1, следует разработать стратегию контроля, обеспечивающую уровень содержания этой примеси в фармацевтической субстанции и лекарственном препарате ниже допустимого предела. Необходимыми условиями разработки надлежащих методов контроля являются знание химических процессов, лежащих в основе синтеза фармацевтической субстанции, и процесса производства лекарственного препарата, а также всестороннее изучение стабильности фармацевтической субстанции и лекарственного препарата.
65. Разработка стратегии контроля мутагенных примесей в лекарственном препарате соотносится с процессами управления рисками, указанными в главе II части III Правил надлежащей производственной практики Евразийского экономического союза, утвержденных Решением Совета Евразийской экономической комиссии от 3 ноября 2016 г. N 77 (далее - Правила производственной практики). Стратегия контроля, основанная на знании фармакологических свойств лекарственного препарата, процессе его производства и сформированная с применением принципов управления рисками, позволяют разработать план контроля процесса производства и надлежащие аналитические испытания, которые обеспечат возможность переноса основного контроля на более ранние стадии технологического процесса и минимизируют необходимость испытаний конечного продукта.
1. Варианты контроля производственных
(технологических) примесей
66. Существуют следующие потенциальные варианты разработки стратегии контроля примесей в составе фармацевтической субстанции.
Вариант 1. Включить испытания на примеси в спецификацию фармацевтической субстанции с критериями приемлемости, равными или меньшими, чем предел допустимого уровня поступления примеси, и с использованием надлежащей аналитической методики.
При таком подходе к контролю возможно применение периодических проверочных испытаний в соответствии с приложением N 1 к Руководству по составлению нормативного документа по качеству лекарственного препарата. Периодические проверочные испытания обоснованы, если можно показать, что содержание мутагенной примеси в фармацевтической субстанции не превышает 30% от допустимого предела приемлемости по меньшей мере для 6 последовательных опытно-промышленных или 3 последовательных промышленных серий лекарственного препарата. Если это условие не выполняется, рекомендуется использовать рутинное испытание по спецификации на фармацевтическую субстанцию.
Вариант 2. Включить в спецификацию сырья, исходного материала, промежуточного продукта или во внутрипроизводственный контроль испытание на примесь с критериями приемлемости, равными или меньшими, чем предел допустимого уровня поступления примеси, и с использованием надлежащей аналитической методики.
Вариант 3. Включить в спецификацию сырья, исходного материала, промежуточного продукта или во внутрипроизводственный контроль испытание на примесь с критериями приемлемости превышающими предел допустимого уровня поступления примеси, с использованием надлежащей аналитической методики, при условии, что имеется возможность получить подтверждение того, что дальнейшее химическое изменение примеси, процедуры очистки от нее и связанные с ней процессы контроля позволят обеспечить в фармацевтической субстанции содержание примеси ниже допустимого уровня ее поступления без необходимости проведения каких-либо дальнейших испытаний.
Этот вариант допускается применять, если содержание примеси в фармацевтической субстанции не превышает 30% от допустимого предела приемлемости, установленного по результатам анализа лабораторных серий лекарственного препарата (рекомендуется также выполнение исследований серий с добавлением примеси) и при необходимости подтвержденного результатами анализа опытно-промышленных или промышленных серий лекарственного препарата. Примеры применения данного варианта приведены в приложении N 2 к настоящему Руководству. Для обоснования приемлемости варианта 3 допускается применять альтернативные подходы.
Вариант 4. Отказаться от аналитических испытаний на содержание примеси и ее включения в спецификацию и перейти на стратегию контроля процесса производства.
Стратегия контроля, основанная на контроле процесса производства вместо выполнения аналитических испытаний, применима в случае, если выполняются все следующие условия:
понятны все химические стадии процесса образования и разрушения примеси;
полностью охарактеризованы параметры производственного процесса и влияние производственного процесса на остаточное содержание мутагенной примеси, в том числе представлены сведения относительно дальнейшего химического изменения примеси и возможности очистки производственной линии от этой примеси;
риск присутствия примеси в лекарственном препарате превышающий допустимый предел приемлемости определен как незначительный.
Как правило, для обоснования этого подхода к контролю следует использовать результаты научных исследований, а также элементы научно обоснованной оценки рисков. Оценку рисков следует выполнять исходя из физико-химических свойств примесей и факторов процесса, которые оказывают влияние на химическое изменение примеси (включая реакционную способность, растворимость, летучесть, ионизируемость примеси) и любые физические воздействия, предназначенные для удаления примесей. Результаты подобной оценки рисков могут быть выражены в качестве расчетного оценочного коэффициента очистки для процесса удаления примеси.
Вариант 4 пригоден для контроля примесей, являющихся нестабильными (например, для тионилхлорида, который быстро и полностью реагирует с водой), или для примесей, которые добавляются на ранних стадиях синтеза и подвергаются в последующем эффективной очистке.
В некоторых случаях подход на основе варианта 4 может применяться в отношении примеси, которая образуется или вводится на поздней стадии синтеза, при этом для обоснования такого подхода следует представить данные, собранные для конкретного процесса.
2. Подходы к контролю производственных
(технологических) примесей
67. В отношении подходов к контролю производственных (технологических) примесей на основе варианта 4 в случае, если обоснование, выполненное только на научных принципах, не считается достаточным, а также в отношении подходов к контролю таких примесей на основе варианта 3 следует представить аналитические данные, обосновывающие выбранный подход к контролю. К ним относятся надлежащие сведения о структурных изменениях примеси, обусловленных последующими стадиями химического синтеза (дальнейшее химическое изменение примеси), результаты анализа опытно-промышленных серий и в некоторых случаях данные исследований опытной серии лекарственного препарата с намеренным добавлением примеси (исследования с добавлением примеси). В этих случаях необходимо подтвердить, что обоснование уровня содержания примеси с учетом ее дальнейшего химического изменения в производственном процессе или очистки производственной линии является надежным и будет всегда обеспечивать присутствие примеси в готовом лекарственном препарате на уровне не превышающем допустимый предел приемлемости.
68. Если коэффициент очистки основан на данных по разработке, необходимо проанализировать возможную зависимость (отсутствие зависимости) от объема промышленных серий лекарственного препарата. Если опытная или опытно-промышленная серия лекарственного препарата, использованная на этапе разработки, не репрезентативна промышленной серии лекарственного препарата, следует представить подтверждение надежности контроля на опытно-промышленных и (или) первых промышленных сериях. На необходимость использования данных опытно-промышленных и (или) промышленных серий влияют:
величина коэффициента очистки, рассчитанная на основе данных лабораторной или опытно-промышленной серии лекарственного препарата;
точка ввода примеси в производственный процесс;
сведения о точках очистки на последующих стадиях производственного процесса.
69. Если проведение контроля примесей на основе вариантов 3 и 4 невозможно обосновать, то следует включить испытание на приемлемое содержание примеси по одному из следующих вариантов:
в спецификацию сырья, исходного материала, промежуточного продукта или во внутрипроизводственный контроль с критериями приемлемости, равными или меньшими, чем предел допустимого уровня поступления примеси (контроль примесей на основе варианта 2);
в спецификацию фармацевтической субстанции с критериями приемлемости, равными или меньшими, чем предел допустимого уровня (контроль примесей на основе варианта 1).
Если не представлено никаких иных обоснований, то контроль примесей, вводимых на последней стадии синтеза фармацевтической субстанции, выполняется на основе варианта 1.
70. Если содержание мутагенной примеси ниже пределов приемлемости, то применение принципа наименьшей практической целесообразности необязательно. Также необязательно подтверждать, что изучались альтернативные пути синтеза.
71. Если методы контроля не позволяют снизить уровень содержания мутагенной примеси ниже допустимого порога приемлемости и такой уровень содержания мутагенной примеси соответствует принципу наименьшей практической целесообразности (основываясь на анализе соотношения "польза - риск"), допускается увеличить предел содержания примеси.
3. Контроль примесей в рамках периодических испытаний
72. Приведенные в подразделах 1 и 2 настоящего раздела варианты и подходы к контролю производственных (технологических) примесей включают в себя случаи, при которых в спецификацию лекарственного препарата рекомендуется включить испытание на оценку содержания примесей, при этом рутинное проведение такого испытания при выпуске каждой серии лекарственного препарата (фармацевтической субстанции) может не требоваться. Подход с проведением испытаний на содержание примесей в рамках периодических испытаний целесообразен, если есть возможность экспериментально подтвердить, что применяемые методы обработки (очистки) после образования (введения) примеси позволяют удалить данную примесь. Следует отметить, что периодические проверочные испытания возможны лишь при использовании процесса, находящегося под контролем (то есть процесса, в результате которого производится качественный лекарственный препарат, удовлетворяющий спецификациям и требованиям надлежащим образом установленного режима контроля производственной площадки, расположенного на ней оборудования, обработки и эксплуатации этого оборудования).
73. Если в ходе испытания на содержание мутагенной примеси результаты не удовлетворяют критериям приемлемости, установленным для периодического испытания, производитель лекарственного препарата должен немедленно начать выполнять полный комплекс испытаний (то есть проводить испытание каждой серии по контролируемому показателю). Такие испытания продолжаются до тех пор, пока не будет установлена причина несоответствия, не будут предприняты корректирующие действия и документально подтверждено, что процесс снова находится под контролем. Уполномоченные органы государств - членов Союза уведомляют о том, что ранее выпущенные серии лекарственного препарата не подвергались такой оценке для выполнения в их отношении повторной оценки соотношения "польза - риск".
4. Контроль продуктов деградации
74. Следует изучить механизм образования потенциального продукта деградации, охарактеризованного как мутагенный для:
процессов производства фармацевтической субстанции и лекарственного препарата;
предполагаемых вариантов упаковки и условий хранения фармацевтической субстанции и лекарственного препарата.
Для определения значимости потенциального продукта деградации следует провести должным образом спланированные ускоренные исследования стабильности (например, при температуре 40 °C и 75% относительной влажности продолжительностью 6 месяцев) в предполагаемой упаковке, используя при этом надлежащие аналитические методики. В качестве альтернативы перед началом долгосрочных исследованиях стабильности в целях установления релевантности пути деградации можно провести должным образом спланированные кинетически эквивалентные краткосрочные ускоренные исследования стабильности при более высоких температурах в предлагаемой упаковке. Этот вид исследований будет необходим для оценки вероятности появления тех потенциальных продуктов деградации, которые не были выявлены в лекарственном препарате и могут образоваться исходя из потенциальных путей его деградации.
75. Если по результатам таких ускоренных исследований ожидается, что образование продукта деградации будет приближаться к допустимому пределу приемлемости в предлагаемой упаковке и условиях хранения, то следует принять меры по контролю образования продукта деградации. В этих случаях при отсутствии иных обоснований следует провести мониторинг продукта деградации в фармацевтической субстанции или лекарственном препарате в рамках основных долгосрочных исследований стабильности в предлагаемых условиях хранения (в предлагаемой упаковке). Пригодность установленного в спецификации предела содержания мутагенного продукта деградации, как правило, зависит от результатов исследований стабильности.
76. Допускается обосновать более высокий порог содержания мутагенного продукта если предполагается, что при данном варианте разработки состава лекарственного препарата и дизайна упаковки невозможно контролировать уровень содержания мутагенного продукта деградации на уровне ниже допустимого предела, и его содержание в то же время соответствует низким уровням (согласно принципу "наименьшей практической целесообразности", основываясь на анализе соотношения "польза - риск").
5. Контроль примесей путем управления жизненным
циклом лекарственного препарата
77. Рекомендации приведенные в настоящем подразделе, применяются в отношении лекарственных препаратов, зарегистрированных после вступления в силу настоящего Руководства.
78. Элементы системы качества и обязанности руководства, описанные в главе III части III Правил производственной практики, допускают использование научных и основанных на анализе рисков подходов к каждому этапу жизненного цикла, способствуя тем самым непрерывному совершенствованию производственного процесса на протяжении всего жизненного цикла лекарственного препарата. Систематизировать и анализировать знания о лекарственном препарате и процессе его производства начинают со стадии разработки и продолжают в течение всего срока обращения лекарственного препарата вплоть до вывода его из обращения, прекращения его производства.
79. Разработка и совершенствование процесса производства фармацевтической субстанции и лекарственного препарата, как правило, продолжаются на протяжении всего их жизненного цикла. Следует регулярно проводить оценку характеристик процесса производства, включая эффективность стратегии контроля. Знания, полученные в ходе промышленного производства, могут использоваться для дальнейшего улучшения понимания процесса производства и его характеристик, а также для корректировки стратегии контроля.
80. Каждое предложение об изменении процесса производства необходимо оценить с точки зрения влияния на качество фармацевтической субстанции и лекарственного препарата. Эта оценка должна основываться на понимании процесса производства, и на ее основе определяется необходимость проведения надлежащих испытаний для анализа влияния предлагаемых изменений. Кроме того, совершенствование аналитических методик может привести к структурной идентификации примеси. В этих случаях новая структура подлежит оценке на предмет мутагенности в соответствии с настоящим Руководством.
81. На протяжении жизненного цикла препарата следует регулярно оценивать необходимость проведения испытаний в случае намеренных или случайных изменений процесса производства, прежде всего при отсутствии рутинного мониторинга допустимых пределов приемлемости (подход к контролю примесей основанный на варианте 3 или 4) или при проведении периодических, а не посерийных испытаний. Такие испытания следует проводить на соответствующей стадии процесса производства.
82. В некоторых случаях использование статистического контроля производственного процесса и отслеживание тенденций изменений производственного процесса целесообразны для подтверждения постоянной пригодности и возможности посредством производственных процессов обеспечивать надлежащий контроль примеси. При статистическом контроле производственного процесса можно использовать параметры процесса, влияющие на образование примеси или очистку от нее, даже если эта примесь не подвергается рутинному мониторингу (например, контролю примесей, основанному на варианте 4).
83. Все изменения проводятся в рамках внутреннего процесса управления изменениями, являющимся частью системы обеспечения качества (глава III части III Правил производственной практики). Об изменениях сведений, содержащихся в регистрационном досье зарегистрированного лекарственного препарата, следует сообщать уполномоченным органам государств - членов Союза.
6. Контроль примесей в рамках клинической
разработки лекарственного препарата
84. Поскольку в ходе клинической разработки лекарственного препарата знания о нем и процессе его производства совершенствуются, предполагается, что данных для обоснования стратегии контроля на этапе клинической разработки будет меньше, чем на стадии регистрации лекарственного препарата. В целях оптимального планирования выполнения аналитического контроля содержания примесей, обладающих наибольшей вероятностью присутствия в фармацевтической субстанции и лекарственном препарате, следует придерживаться подхода, основанного на оценке риска, используя знания химико-технологических процессов производства фармацевтической субстанции и лекарственного препарата. При обосновании ранних этапов клинической разработки лекарственного препарата, когда вероятность присутствия примеси в нем низкая, аналитические данные относительно нормирования уровня примеси могут отсутствовать. Однако получение таких аналитических данных целесообразно для обоснования подхода к контролю, описанному в регистрационном досье лекарственного препарата. Поскольку подбор состава и способа производства промышленных серий лекарственного препарата производится на более поздних этапах клинической разработки, возможность контроля примесей, обусловленных продуктами деградации лекарственного препарата, на ранних фазах разработки может быть ограничена.
IX. Документация по оценке и контролю ДНК-реактивных
(мутагенных) примесей лекарственных средств
1. Документация в досье на клинические
исследования лекарственного препарата
85. На протяжении периода клинической разработки лекарственного препарата количество соединений, проверенных на мутагенность, и объем аналитических данных относительно мутагенных свойств этих соединений должны увеличиваться.
86. Для клинических исследований I фазы продолжительностью 14 дней или меньше следует включить описание действий по уменьшению рисков мутагенности, связанных с примесями классов 1 и 2, а также примесями, перечисленными в разделе VII настоящего Руководства, входящими в группу, вызывающую опасения. Для клинических исследований I фазы, продолжительностью более 14 дней, и клинических исследований IIa фазы необходимо дополнительно включить изучение примесей класса 3, требующих аналитических испытаний.
87. Для клинических исследований IIb и III фаз необходимо включить перечень примесей, оцененных на предмет количественной структурно-функциональной зависимости, а также описать любые фактические и потенциальные примеси классов 1, 2 и 3 вместе с планами их контроля. Следует описать выбранные in silico системы количественной структурно-функциональной зависимости, использованные для проведения оценки. Следует представить результаты испытаний фактических примесей на мутагенность в бактериальных тест-системах. Взамен аналитических данных потенциальных примесей, которые могут присутствовать с низкой вероятностью, следует применять положения указанные в подразделе 6 раздела VIII настоящего Руководства.
2. Документация в составе регистрационного досье
лекарственного препарата в формате общего
технического документа
88. Для фактических и потенциальных примесей и продуктов разложения, связанных с процессом производства, оценка которых проводится в соответствии с настоящим Руководством, следует предоставить классификацию мутагенных примесей и обоснование этой классификации, включающее в себя:
результаты и описание использованных in silico систем количественной структурно-функциональной зависимости, а также вспомогательные сведения для составления общего заключения по обоснованию уровней примесей классов 4 и 5;
отчеты об исследованиях мутагенности, если примеси изучены в тестах на мутагенность у бактерий.
Следует представить обоснование предлагаемой спецификации и подхода к контролю. Например, эти сведения могут включать информацию о допустимом проведении, расположении и чувствительности рутинного мониторинга. В отношении подходов к контролю, основанных на вариантах 3 и 4, необходимо представить обзор данных о коэффициенте очистки и идентификации факторов, обеспечивающих контроль уровня содержания примеси (например, стадии процесса появления примеси, растворимость примеси в промывающих растворах).
Приложение N 1
к Руководству по оценке
и контролю ДНК-реактивных (мутагенных)
примесей в лекарственных средствах
и установлению границ потенциального
канцерогенного риска
ПОДХОДЫ
К ПРИМЕНЕНИЮ РУКОВОДСТВА ПО ОЦЕНКЕ И КОНТРОЛЮ
ДНК-РЕАКТИВНЫХ (МУТАГЕННЫХ) ПРИМЕСЕЙ В ЛЕКАРСТВЕННЫХ
СРЕДСТВАХ И УСТАНОВЛЕНИЮ ГРАНИЦ ПОТЕНЦИАЛЬНОГО
КАНЦЕРОГЕННОГО РИСКА
Варианты
Оценка фармацевтической субстанции
Оценка лекарственного препарата
Комментарии
1
2
3
4
I. Регистрация лекарственных препаратов и внесение изменений в регистрационное досье
1. Лекарственный препарат из новой (ранее не разрешенной к медицинскому применению) фармацевтической субстанции
да
да
полное применение Руководства но оценке и контролю ДНК-реактивных (мутагенных) примесей в лекарственных средствах и установлению границ потенциального канцерогенного риска (далее - Руководство)
2. Лекарственный препарат из разрешенной к медицинскому применению фармацевтической субстанции с новым составом вспомогательных веществ
нет
да
подраздел 2 раздела IV Руководства
3. Оригинальный лекарственный препарат подан на процедуру приведения регистрационного досье в соответствие с Правилами регистрации и экспертизы лекарственных средств для медицинского применения, утвержденными Решением Совета Евразийской экономической комиссии от 3 ноября 2016 г. N 78. При этом лекарственный препарат не изменялся
да
да
рассматривается в качестве лекарственного препарата, изготовленного из новой фармацевтической субстанции с точки зрения изучения мутагенности
4. В регистрационное досье зарегистрированного лекарственного препарата вносится новый поставщик фармацевтической субстанции или новая площадка производства фармацевтической субстанции. Изменения процесса производства при этом отсутствуют
нет
нет
переоценка риска мутагенных примесей не требуется, если синтез фармацевтической субстанции соответствует ранее одобренным методам. Заявителю при этом необходимо подтвердить, что изменения ранее одобренного процесса производства или состава препарата не произошло (подраздел 1 раздела IV Руководства)
5. В регистрационное досье зарегистрированного лекарственного препарата с повышенными пределами ДНК-реактивных примесей, применяющегося при лечении распространенного рака, вносится дополнительное показание для применения по не угрожающему жизни состоянию
да
да
поскольку популяция пациентов и приемлемый канцерогенный риск изменились, ранее одобренная стратегия контроля примесей и установленные пределы содержания примесей потребуют переоценки (подраздел 3 раздела IV Руководства)
6. Комбинированный лекарственный препарат, содержащий хотя бы 1 новую фармацевтическую субстанцию и известные фармацевтические субстанции
да
(для новой фармацевтической субстанции)
да
Руководство применяется к новой фармацевтической субстанции. Руководство не применяется к известной фармацевтической субстанции. Лекарственный препарат будет рассматриваться в качестве нового, поэтому Руководство будет применяться ко всем новым продуктам деградации или повышенному содержанию ранее имевшихся примесей
нет
(для известной фармацевтической субстанции)
III. Заявление на разрешение клинических исследований
7. Лекарственный препарат из новой (ранее не разрешенной к медицинскому применению) фармацевтической субстанции
да
да
полное применение Руководства
8. Новая фармацевтическая субстанция для производства препаратов противоопухолевой терапии
нет
нет
вне сферы применения Руководства
9. Новая фармацевтическая субстанция для производства орфанных препаратов
да
да
могут быть индивидуальные исключения по применению Руководства для более высоких пределов содержания примесей
10. Новый лекарственный препарат с использованием известной фармацевтической субстанции при отсутствии изменений процесса производства последней
нет
да
в отсутствие изменений синтеза фармацевтической субстанции ретроспективное применение Руководства в отношении этой субстанции не предполагается. Фармацевтическая субстанция не требует переоценки. Поскольку лекарственный препарат при этом новый, в его отношении следует применять положения Руководства
Приложение N 2
к Руководству по оценке
и контролю ДНК-реактивных (мутагенных)
примесей в лекарственных средствах
и установлению границ потенциального
канцерогенного риска
ПРИМЕРЫ
ПРИМЕНЕНИЯ ПОТЕНЦИАЛЬНЫХ ПОДХОДОВ К КОНТРОЛЮ ДНК-РЕАКТИВНЫХ
(МУТАГЕННЫХ) ПРИМЕСЕЙ В ЛЕКАРСТВЕННЫХ СРЕДСТВАХ В ЦЕЛЯХ
СНИЖЕНИЯ ПОТЕНЦИАЛЬНОГО КАНЦЕРОГЕННОГО РИСКА
1. Пример применения стратегии контроля,
основанной на варианте 3
Промежуточный продукт X образуется за 2 стадии производственного процесса до получения фармацевтической субстанции, и в продукте X рутинно обнаруживается примесь A. Примесь A является стабильным соединением и переносится в фармацевтическую субстанцию. В лабораторных условиях было проведено исследование с добавлением различных концентраций примеси A к промежуточному продукту X. В результате этих исследований установлено стабильное удаление примеси A из фармацевтической субстанции до менее чем 30% от предела порога токсикологической угрозы, даже если содержание исходное содержание примеси A в промежуточном продукте X составляло 1%.
Поскольку промежуточный продукт X образуется за 2 стадии производственного процесса до получения фармацевтической субстанции, а содержание примеси A в нем относительно высокое, очищающая способность процесса была дополнительно подтверждена определением содержания примеси A в фармацевтической субстанции в нескольких опытно-промышленных сериях. Результаты показали содержание примеси в субстанции ниже 30% от предела порога токсикологической угрозы. Таким образом, контроль содержания примеси A в промежуточном продукте X с допустимым критерием приемлемости 1,0% обоснован, поэтому включение испытания на содержание этой примеси в спецификацию на фармацевтическую субстанцию не требуется.
2. Пример применения стратегии контроля, основанной
на варианте 3, при прогнозируемой очистке по результатам
исследования с добавлением примеси и использованием
стандартных аналитических методов
Исходный материал Y вносится на 3-й стадии 5-стадийного синтеза, в котором с помощью стандартных аналитических методов рутинно обнаруживается примесь B в количестве менее 0,1%. В целях определения критерия приемлемости содержания примеси на уровне 0,1% в спецификации на исходный материал, было проведено лабораторное исследование очистки, в ходе которого примесь B добавлялась к исходному материалу Y в различных концентрациях (до 10%), коэффициент очистки на 3 конечных стадиях обработки составил менее 500. Применение этого коэффициента очистки в отношении предела содержания примеси на уровне 0,1% в спецификации на исходный материал Y дает прогнозируемое содержание примеси B в фармацевтической субстанции, которое равно менее 2 ppm. Поскольку это значение ниже предела токсикологической угрозы, равного 50 ppm для этой примеси в фармацевтической субстанции, предела содержания примеси B на уровне 0,1% в спецификации на исходный материал Y обоснован без необходимости представления результатов анализа опытно-промышленных или промышленных серий фармацевтической субстанции.
3. Пример применения подхода к контролю,
основанному на вариантах 2 и 4, при структурно
схожих мутагенных примесях
Промежуточный продукт 1-й стадии 5-стадийного синтеза является нитроароматическим соединением, которое может содержать низкое количество примеси C (позиционного изомера промежуточного продукта 1 стадии, представляющего собой также нитроароматическое соединение). Количество примеси C в промежуточном продукте 1-й стадии с помощью стандартных аналитических методов не изучалось, но она может присутствовать в меньшем количестве. Промежуточный продукт 1-й стадии имеет положительную реакцию в тесте на мутагенность у бактерий. Реакция гидрогенизации 2-й стадии приводит к 99%-ной конверсии промежуточного продукта 1-й стадии в соответствующий ароматический амин. Это подтверждается внутрипроизводственным испытанием.
Была проведена оценка очистки оставшегося нитроароматического промежуточного продукта 1-й стадии, и был спрогнозирован высокий коэффициент очистки, в точках очистки на последующих 3-й и 4-й стадиях процесса обработки. Очистка на 5-й стадии обработки не предполагается, поэтому спецификация на промежуточный продукт 1-й стадии с пределом на основе порога токсикологической угрозы была установлена для промежуточного продукта 4-й стадии (вариант 2 подхода к контролю). Очистка от позиционно изомерной примеси C ожидается на тех же точках очистки, что и в случае промежуточного продукта 1-й стадии, поэтому ее содержание всегда будет намного ниже содержания самого промежуточного продукта 1-й стадии, поэтому испытания не требуются, а вариант 4 подхода контроля примеси C может быть обоснован без необходимости дополнительных лабораторных или опытно-промышленных данных.
4. Пример применения стратегии контроля, основанной
на варианте 4, при высокореактивной примеси
Тионилхлорид является высокореактивным мутагенным соединением. Например, если тионилхлорид вносится на 1-й стадии 5-стадийного синтеза и на различных стадиях этого процесса синтеза используется большое количество воды, то поскольку тионилхлорид мгновенно вступает в реакцию с водой, вероятность его остаточного присутствия в фармацевтической субстанции нулевая. Таким образом, в данном случае, пригоден вариант 4 подхода к контролю без необходимости представления каких-либо дополнительных лабораторных или промышленных данных по контролю остаточных количеств тионилхлорида.