"МР 2.6.1/2.3.7.0216-20. 2.6.1. Ионизирующее излучение, радиационная безопасность. 2.3.7. Состояние здоровья населения в связи с состоянием питания. Радиохимическое определение удельной активности природных радионуклидов в пробах пищевой продукции, почвы,
ГОСУДАРСТВЕННОЕ САНИТАРНО-ЭПИДЕМИОЛОГИЧЕСКОЕ НОРМИРОВАНИЕ
РОССИЙСКОЙ ФЕДЕРАЦИИ
Утверждаю
Руководитель Федеральной
службы по надзору в сфере
защиты прав потребителей
и благополучия человека,
Главный государственный
санитарный врач
Российской Федерации
А.Ю.ПОПОВА
22 сентября 2020 г.
2.6.1. ИОНИЗИРУЮЩЕЕ ИЗЛУЧЕНИЕ, РАДИАЦИОННАЯ БЕЗОПАСНОСТЬ
2.3.7. СОСТОЯНИЕ ЗДОРОВЬЯ НАСЕЛЕНИЯ В СВЯЗИ
С СОСТОЯНИЕМ ПИТАНИЯ
РАДИОХИМИЧЕСКОЕ ОПРЕДЕЛЕНИЕ УДЕЛЬНОЙ АКТИВНОСТИ ПРИРОДНЫХ
РАДИОНУКЛИДОВ В ПРОБАХ ПИЩЕВОЙ ПРОДУКЦИИ, ПОЧВЫ, ДРУГИХ
ОБЪЕКТОВ ОКРУЖАЮЩЕЙ СРЕДЫ И БИОПРОБАХ
МЕТОДИЧЕСКИЕ РЕКОМЕНДАЦИИ
МР 2.6.1/2.3.7.0216-20
1. Разработаны: ФБУН "Санкт-Петербургский научно-исследовательский институт радиационной гигиены имени профессора П.В. Рамзаева" Роспотребнадзора (Кадука М.В., Басалаева Л.Н., Бекяшева Т.А., Иванов С.А., Салазкина Н.В., Ступина В.В.).
2. Утверждены Руководителем Федеральной службы по надзору в сфере защиты прав потребителей и благополучия человека, Главным государственным санитарным врачом Российской Федерации А.Ю. Поповой 22 сентября 2020 г.
3. Введены впервые.
I. ОБЛАСТЬ ПРИМЕНЕНИЯ
1.1. Настоящие методические рекомендации (далее - МР) распространяются на порядок приготовления счетных образцов и определения удельной активности природных радионуклидов 226Ra, 224Ra, 228Ac(228Ra), 238U, 234U, 235U, 210Po и 210Bi(210Pb) в пробах пищевых и сельскохозяйственных продуктов, почвы, других объектов окружающей среды и биопробах с применением радиохимической подготовки проб.
1.2. МР обеспечивают единые подходы при выполнении исследований удельной активности природных радионуклидов в пробах пищевых и сельскохозяйственных продуктов, почвы, других объектов окружающей среды и биопробах для целей радиационно-гигиенической паспортизации, социально-гигиенического мониторинга и единой системы контроля индивидуальных доз (далее - ЕСКИД).
1.3. МР предназначены для органов и организаций Федеральной службы по надзору в сфере защиты прав потребителей и благополучия человека, а также могут быть использованы организациями, осуществляющими радиационный контроль пищевых и сельскохозяйственных продуктов, почвы, других объектов окружающей среды и биопроб, в том числе для оценки доз облучения населения от природных радионуклидов.
II. ОБЩИЕ ПОЛОЖЕНИЯ
2.1. Основным источником поступления в организм человека природных радионуклидов (далее - ПРН) являются пищевые продукты. Содержание радионуклидов в пищевых продуктах зависит от их активности и форм нахождения в объектах окружающей среды, а также природных условий, определяющих миграцию изучаемых радионуклидов по пищевым цепям, технологии переработки сельскохозяйственной продукции и изготовления пищевых продуктов.
2.2. Содержание ПРН в пищевых продуктах дает вклад в суммарную дозу облучения населения, хотя он и не является преобладающим. В отдельных случаях вклад в дозу внутреннего облучения за счет содержания ПРН в пищевых продуктах может быть значительным.
2.3. Основными дозообразующими радионуклидами, наряду с изотопами урана (238U и 234U), являются изотопы радия (226Ra, 228Ra и 224Ra). 226Ra имеет высокий индекс токсичности, связанный с долговременным отложением в костной ткани, высокими энергиями альфа и бета-излучения самого 226Ra и его дочерних продуктов распада. Свинец 210Pb и полоний 210Po вносят существенный, а иногда и основной вклад в естественный радиационный фон, воздействующий на различные биологические объекты, включая человека. 210Pb и его дочерний продукт 210Po по радиотоксикологическому действию на организм отнесены к группе радионуклидов с особо высокой токсичностью.
III. ПРИНЦИПЫ ОПРЕДЕЛЕНИЯ УДЕЛЬНОЙ АКТИВНОСТИ ПРИРОДНЫХ
РАДИОНУКЛИДОВ В ПРОБАХ ПИЩЕВЫХ ПРОДУКТОВ, ПОЧВЫ, ДРУГИХ
ОБЪЕКТОВ ОКРУЖАЮЩЕЙ СРЕДЫ И БИОПРОБАХ
3.1. Диапазон и относительная погрешность измерений радиологических показателей проб пищевых и сельскохозяйственных продуктов, почвы, других объектов окружающей среды и биопроб приведены в таблице 1.
Таблица 1
Диапазон и относительная погрешность
измерений радиологических показателей проб пищевых
и сельскохозяйственных продуктов, почвы, других объектов
окружающей среды и биопроб
|
Диапазон измерений, Бк/пробу |
Относительная погрешность, % (P = 0,95) |
|
свыше 0,02 |
50 |
|
свыше 0,1 |
40 |
|
свыше 0,5 |
30 |
|
свыше 1,0 |
20 |
3.2. Из отобранной пробы исследуемого объекта изготавливают счетные образцы с применением методики радиохимического выделения радионуклидов. Для определения содержания природных радионуклидов в пробах пищевой и сельскохозяйственной продукции и объектов окружающей среды отбирают 0,5 - 1,5 кг образца, в пробах почвы и биопробах - от 0,1 до 0,5 кг.
3.3. Перед озолением пробы пищевого продукта следует провести предварительную пробоподготовку, аналогичную подготовке конкретного вида пищевого продукта перед кулинарной обработкой: очистка от загрязнений, косточек, кожуры, мытье, удаление костей для проб мяса и рыбы и другие виды подготовки, характерные для конкретного пищевого продукта. Пробы объектов окружающей среды очищают от песка, почвы, посторонних примесей. Пробы почвы высушивают до воздушно-сухого веса. Специальной подготовки биопроб не требуется.
3.4. Радиохимическое выделение изотопов радия и урана в озоленной пробе основано на переводе данных радионуклидов в растворимое состояние путем кислотной (HCl) и содовой (Na2CO3) обработки (выщелачивания) пробы.
3.5. Радиохимическое выделение изотопов полония и свинца основано на мокром озолении высушенной пробы с применением концентрированных азотной и соляной кислот с добавлением перекиси водорода. Прием мокрого озоления применяется в данном случае ввиду летучести соединений полония.
3.6. Методика определения удельной активности изотопов радия в пробах включает в себя следующие операции: концентрирование изотопов радия методом осаждения на сульфате бария, отделение от мешающих изотопов, селективное разделение. Методика предусматривает осаждение изотопов радия из раствора пробы вместе с носителем (хлористый барий) серной кислотой в форме BaSO4, который является счетным образцом для измерения активности 226Ra и 224Ra. Активность 228Ra определяют по дочернему 228Ac, который осаждают совместно с носителем (хлористый лантан) в виде La(OH)3. После дальнейшего прокаливания полученный La2O3(228Ac) служит счетным образцом для измерения активности 228Ac(228Ra). Активность счетных образцов, полученных после радиохимического выделения радионуклидов, измеряют с использованием малофонового альфа-бета радиометра.
3.7. Метод определения удельной активности альфа-излучающих изотопов урана основан на их хроматографическом выделении на ионобменной колонке с использованием полифункционального низкоосновного анионита конденсационного типа ЭДЭ-10П с последующим электролитическим осаждением на диск из нержавеющей стали. Активность полученного счетного образца измеряют с использованием полупроводникового альфа-спектрометра.
3.8. Метод определения удельной активности полония и свинца основан на электрохимическом осаждении радионуклидов на никелевом диске из солянокислых растворов (электролитическое замещение или бестоковый электролиз). Определение 210Pb проводят по дочернему 210Bi, который так же, как и 210Po, электрохимически осаждается на никелевом диске. Активность счетного образца, полученного после радиохимического выделения радионуклидов, измеряют с использованием малофонового альфа-бета радиометра.
3.9. Для контроля неизбежных в процессе анализа потерь радионуклидов в исследуемую пробу на первом этапе ее обработки для каждого из определяемых изотопов добавляют известное количество носителя, в качестве которого используют солянокислые или азотнокислые соли соответствующих элементов.
IV. СРЕДСТВА ИЗМЕРЕНИЙ, ВСПОМОГАТЕЛЬНОЕ ОБОРУДОВАНИЕ
И РЕАКТИВЫ И МАТЕРИАЛЫ
Измерительное оборудование
4.1. Альфа-спектрометр со следующими характеристиками:
- время установления рабочего режима - не более 10 мин;
- интегральная нелинейность в диапазоне энергий 4 ч ![]() 9 МэВ - не более 0,1%;
9 МэВ - не более 0,1%;
- энергетическая ширина канала - не более 5,0 кэВ;
- эффективность регистрации излучения радионуклида 239Pu не менее 0,25 Бк-1·с-1 на расстоянии 5 мм от детектора;
- временная нестабильность характеристики преобразования - не более 2% за 8 часов измерений;
- абсолютное энергетическое разрешение по энергиям 5,16 МэВ, 5,49 МэВ и 5,81 МэВ - менее 50 кэВ.
4.2. Альфа-бета радиометр малофоновый с характеристиками не хуже следующих:
- скорость счета фона - не более 0,05 имп/мин по альфа-каналу и не более 2,3 имп/мин - по бета-каналу;
- эффективность регистрации бета-излучения - не ниже 7,5 имп/(мин-Бк);
- эффективность регистрации альфа-излучения электрохимических счетных образцов с диаметром активного пятна 26 мм - не ниже 16,2 имп/(мин·Бк);
- основная погрешность определения активности счетных образцов - не более 10% при P = 0,95.
4.3. Эталонные радионуклидные источники и растворы радионуклидов:
- образцовый спектрометрические источники альфа-излучения 2-го разряда, активностью в диапазоне 100 - 5 000 Бк;
- образцовый радиометрический источник альфа-излучения 2-го разряда активностью в диапазоне 10 - 100 Бк;
- образцовый радиометрический источник бета-излучения 2-го разряда активностью в диапазоне 10 - 100 Бк;
- эталонные растворы радионуклидов 226Ra, 232U, (90Sr + 90Y) объемной активностью от 3 до 25 Бк/см3 с погрешностью измерений объемной активности не более 6% (в случае, если активность эталонного раствора выше указанной, его разбавляют);
- контрольный образец сравнения типа ОСК-210 (210Po + 210Bi), активностью от 10 до 50 Бк с погрешностью измерений активности не более 6%.
4.4. Средства измерения массы и объема, применяемые при приготовлении счетных образцов:
- весы аналитические;
- весы технические;
- пипетки мерные 2-го класса на 1,0, 2,0 и 10 мл;
- колбы мерные 2-го класса на 100 и 250 мл;
- стаканы лабораторные на 100, 200, 800 и 1000 мл.
4.5. Допускается применение других средств измерений, характеристики которых не хуже перечисленных в пп. 4.1 - 4.4.
Все средства измерений должны иметь действующие свидетельства о метрологической поверке.
Вспомогательное оборудование
4.6. Вспомогательное оборудование:
- плитка электрическая;
- дистиллятор;
- печь муфельная электрическая с терморегулятором до 900 °C;
- колонка хроматографическая стеклянная с внутренним диаметром 10 мм;
- форвакуумный насос, обеспечивающий рабочее давление в вакуумной камере не более 1333 Па (10 мм рт. ст.);
- разборная установка для электролитического осаждения радионуклидов на подложки из нержавеющей стали, включающая электролитическую ячейку с анодным и катодным электродами;
- регулируемый источник питания постоянного тока, обеспечивающий максимальный ток до 3 А и максимальное напряжение до 30 В;
- никелевые диски диаметром 34 мм, изготовленные из никелевой ленты;
- наждачная бумага мелкозерновая;
- индикаторная бумага универсальная pH 1 - 10;
- чашки и тигли фарфоровые на 25, 50 и 100 мл;
- ступка фарфоровая;
- стекло часовое;
- палочки стеклянные диаметром 3 мм;
- шпатель;
- воронки стеклянные диаметром 5, 7,5, 10 и 15 см;
- колба круглодонная;
- колба коническая;
- подложки из нержавеющей стали;
- тефлоновая кассета для электролитического осаждения;
- бумага фильтровальная;
- перчатки резиновые;
- вата.
4.7. Допускается применение другого вспомогательного оборудования, характеристики которого не хуже перечисленных в п. 4.6.
Реактивы и материалы
4.8. Реактивы:
- аммиак водный NH4OH;
- аммоний углекислый (NH4)CO3;
- аммоний хлористый NH4Cl;
- аммоний щавелевокислый (NH4)2C2O4;
- барий хлористый BaCl2;
- бария гидроокись Ba(OH)2;
- вода дистиллированная H2O;
- железо хлорное 6-водное FeCl3·6H2O;
- иттрий хлористый 6-водный YCl3·6H2O;
- калий хлористый KCl;
- карбонат натрия (сода) Na2CO3;
- кислота азотная концентрированная HNO3;
- кислота аскорбиновая;
- кислота лимонная 1-водная;
- кислота серная концентрированная H2SO4;
- кислота соляная концентрированная HCl;
- лантан хлористый 7-водный LaCl3·7H2O;
- перекись водорода 30% H2O2;
- спирт этиловый C2H5OH;
- трилон Б Na4-EDTA;
- уранил азотнокислый UO2(NO3)2·6H2O;
- анионообменная смола ЭДЭ-10П или аналогичная по ионообменным характеристикам, размер зерен 0,125 - 0,250 мм;
4.9. Все используемые для выполнения анализа реактивы и материалы должны быть марок х.ч. или ч.д.а. Процедура приготовления рабочих растворов и растворов носителей приведена в приложении 1 к настоящим МР. Процедура приготовления калибровочных образцов и градуировки альфа-бета радиометра приведена в приложении 2 к настоящим МР.
V. ТРЕБОВАНИЯ К БЕЗОПАСНОСТИ И КВАЛИФИКАЦИИ ПЕРСОНАЛА
5.1. Работы должны проводиться с соблюдением требований инструкции по технике безопасности при работе с вредными веществами.
5.2. При работе с измерительными приборами и вспомогательным оборудованием необходимо соблюдать требования безопасной эксплуатации, изложенные в соответствующих разделах технической документации на приборы.
5.3. К выполнению работ допускаются лица не моложе 18 лет, прошедшие практическую подготовку по соответствующей программе, имеющие квалификацию химика, оператора (лаборанта) по радиометрическим и спектрометрическим измерениям, врача по санитарно-гигиеническим лабораторным исследованиям, врача-лаборанта, фельдшера-лаборанта или инженерно-технические работники, допущенные к выполнению работ в установленном порядке, ознакомленные с руководством по эксплуатации и техническим описанием альфа-бета радиометра и/или альфа-спектрометра.
VI. РАДИОХИМИЧЕСКОЕ ВЫДЕЛЕНИЕ 226Ra, 224Ra, 228Ac(228Ra)
И ПРИГОТОВЛЕНИЕ СЧЕТНЫХ ОБРАЗЦОВ ДЛЯ ОПРЕДЕЛЕНИЯ
ИХ УДЕЛЬНОЙ АКТИВНОСТИ
6.1. Подготовленную согласно п. 3.3 пробу твердых образцов взвешивают (вес записывают в рабочем журнале), измельчают, высушивают при комнатной температуре, помещают в металлическую (фарфоровую) посуду, сжигают на электроплитке и озоляют до серого цвета в муфельной печи при температуре 500 - 600 °C для удаления органики. Жидкие, предварительно взвешенные пробы, выпаривают в термостойких стаканах или фарфоровой посуде до минимального объема, затем также сжигают и озоляют до серого цвета в муфельной печи.
6.2. Озоленную пробу помещают в стакан емкостью 200 мл, вносят растворы носителей: хлорид бария (90 - 100 мг в расчете на BaSO4) и хлорид железа (30 мг в расчете на Fe2O3, добавляют изотопный индикатор 232U, активность которого не должна превышать ожидаемых в пробе активностей изотопов урана более чем в 5 - 10 раз, как правило, в пробу вносят 0,2 - 0,3 Бк 232U. Пробу заливают 100 - 200 мл 6 H HCl и кипятят 1,5 - 2 часа при периодическом помешивании стеклянной палочкой. Раствор отфильтровывают, используя обеззоленный фильтр "белая лента" 12,5 см (фильтрат 1).
6.3. Для полного перевода в раствор радионуклидов радия и урана после фильтрования нерастворившийся осадок переносят дистиллированной водой в стакан емкостью 200 мл, добавляют 5 - 7 г Na2CO3, доводят до 100 - 120 мл и кипятят в течение 1,5 часов, периодически добавляя воду до первоначального объема. Полученный осадок карбонатов радия-бария отфильтровывают, используя обеззоленный фильтр "синяя лента" 12,5 см, промывают горячей дистиллированной водой, растворяют в 2 H HCl и объединяют с основным фильтратом (фильтрат 1). Карбонатный содовый раствор кипятят с добавлением соляной кислоты до полного удаления CO2 и объединяют с фильтратом 1 непосредственно перед осаждением сульфатов радия-бария.
6.4. Объединенный раствор разбавляют дистиллированной водой до pH 1 - 1,5 (предполагаемый объем 800 - 1000 мл), доводят до кипения, добавляют при непрерывном перемешивании 40 мл 10% серной кислоты и кипятят (после появления осадка) при медленном нагревании еще 10 - 15 минут, после чего дают осадку отстояться 3 - 4 часа (лучше оставить на ночь).
6.5. После отстаивания осадок сульфатов (бария-радия) отфильтровывают через обеззоленный фильтр "синяя лента" 12,5 см, промывают дистиллированной водой и затем переносят осадок с помощью 70 - 100 мл дистиллированной воды в стакан объемом 150 мл. Фильтрат (фильтрат 2) оставляют для определения содержания в пробе изотопов урана.
6.6. К осадку сульфатов (бария-радия) добавляют 5 - 7 г соды (Na2CO3) для перевода сульфатов в карбонаты и кипятят в течение 2-х часов при периодическом помешивании, подливая время от времени дистиллированную воду до объема раствора 100 мл. Осадок карбонатов отфильтровывают, используя обеззоленный фильтр "синяя лента" 12,5 см, промывают горячей дистиллированной водой и растворяют в 2 H HCl.
6.7. Полученный раствор кипятят до полного удаления CO2. После чего в пробу вносят раствор хлорного железа (10 мг в расчете на Fe2O3) и осаждают безугольным аммиаком гидроокись железа для дополнительной очистки радия от сопутствующих радионуклидов. Осадок отфильтровывают, используя обеззоленный фильтр "красная лента" 12,5 см и промывают горячей дистиллированной водой и отбрасывают.
6.8. Фильтрат, полученный после отделения осадка гидроокиси железа, и содержащий радиохимически чистые изотопы радия, подкисляют до pH = 2, затем вносят раствор носителя лантана (60 мг в расчете на La2O3) и оставляют на 36 - 40 часов для накопления 228Ac из 228Ra.
6.9. После накопления 228Ac раствор кипятят для удаления CO2, после чего осаждают гидроокись лантана (актиния) безугольным аммиаком. Осадок отфильтровывают через обеззоленный фильтр "синяя лента" 12,5 см (время окончания фильтрования, т.е. время отделения 228Ac от 228Ra, записывают), промывают горячей дистиллированной водой с добавлением безугольного аммиака, высушивают, прокаливают при температуре 800 °C, растирают в тигле стеклянной палочкой до состояния "пудры", переносят на предварительно взвешенную подложку, фиксируют несколькими каплями этилового спирта, равномерно распределяют и высушивают. После высушивания подложку с осадком снова взвешивают для расчета веса выделенного осадка и определения по нему химического выхода.
6.10. Измерение активности 228Ac в приготовленном счетном образце и определение его удельной активности в пробе выполняют в соответствии с главой 10 настоящих МР.
6.11. Фильтрат (после отделения 228Ac на осадке гидроокиси лантана) подкисляют HCl до pH = 1, нагревают до кипения и осаждают сульфат бария (радия), добавив 10 мл 10% раствора H2SO4.
6.12. После отстаивания осадок отфильтровывают, используя обеззоленный фильтр "синяя лента" диаметром 12,5 см, промывают дистиллированной водой, высушивают и прокаливают при температуре 800 °C в течение 1 часа. Записывают время окончания прокаливания. Осадок растирают, равномерно наносят на предварительно взвешенную подложку, фиксируют несколькими каплями этилового спирта, равномерно распределяют и высушивают. После высушивания подложку с осадком снова взвешивают для расчета веса выделенного осадка и определения по нему химического выхода.
6.13. Измерение активности 224Ra и 226Ra в приготовленном счетном образце и определение их удельной активности в пробе выполняют в соответствии с главой 10 настоящих МР.
VII. РАДИОХИМИЧЕСКОЕ ВЫДЕЛЕНИЕ 238U, 234U, 235U
И ПРИГОТОВЛЕНИЕ СЧЕТНЫХ ОБРАЗЦОВ ДЛЯ ОПРЕДЕЛЕНИЯ
ИХ УДЕЛЬНОЙ АКТИВНОСТИ
7.1. Фильтрат 2 после отделения сульфатов бария-радия кипятят в стакане емкостью 800 мл не менее 30 минут для удаления CO2. Затем безугольным аммиаком при pH = 9 осаждают гидроокиси железа, содержащие изотопы урана, тория и редкоземельных элементов. Осадок отфильтровывают через обеззоленный фильтр "красная лента" диаметром 12 - 15 см, промывают 2 - 3 раза горячей дистиллированной водой и растворяют в 2 H HCl. Полученный раствор разбавляют до объема 500 - 700 мл дистиллированной водой, добавляют 10 - 15 мл карбоната аммония и при нагревании вновь осаждают гидроокиси железа, содержащие изотопы урана, тория и редкоземельных элементов (pH = 8 - 9), используя раствор аммиака до полной коагуляции осадка.
7.2. После отстаивания осадок отфильтровывают через обеззоленный фильтр "белая лента" диаметром 12 - 15 см и промывают 2 - 3 раза дистиллированной водой. Данная операция необходима для отделения соединений урана от соединений тория и редкоземельных элементов. Осадок оставляют для определения изотопов тория, либо отбрасывают. К фильтрату, содержащему карбонатный комплекс урана, добавляют 10 - 15 мл смеси концентрированных HCl и HNO3 в соотношении 1:1 и упаривают его до влажных солей.
7.3. К влажным солям карбонатного комплекса урана добавляют минимальный объем (10 - 15 мл) смеси концентрированных HCl и HNO3 (1:1) и медленно упаривают до образования влажных солей. Эту операцию повторяют несколько раз для полного разрушения карбонатного комплекса урана и удаления белого окрашивания осадка аммонийных солей.
7.4. К обработанным в соответствии с п. 9.3 влажным солям урана добавляют минимальный объем (10 - 15 мл) концентрированной HCl и медленно упаривают до влажных солей для удаления HNO3. Данную операцию повторяют 2 - 3 раза до прекращения выделения из раствора бурых паров оксидов азота. Прекращение выделения бурых паров будет свидетельствовать о полном удаление HNO3 из образца.
7.5. Обработанный упаренный остаток солей урана растворяют в 50 мл 8 H HCl. Полученный раствор пропускают через колонку диаметром 10 мм с 15 мл анионообменной смолы, подготовленной согласно приложению 1 к настоящим МР, со скоростью 1 мл/мин. Затем через колонку пропускают 70 мл 8 H HCl. Объединенные элюаты, содержащие изотопы тория, оставляют для дальнейшего определения этих радионуклидов или отбрасывают.
7.6. Изотопы урана элюируют с колонки 0,5 H HCl. Объем элюата должен составлять 100 - 150 мл, что обеспечивает полноту десорбции урана. Полученный раствор упаривают до влажных солей.
7.7. К влажным солям урана добавляют 5 - 10 мл концентрированной HCl и упаривают их до сухого остатка. К полученному сухому остатку добавляют 10 мл 0,5 H HNO3 и, при необходимости (если сухой остаток не растворяется), нагревают. Затем в полученный раствор вносят 5 мл 1% раствора трилона Б, 1 мл насыщенного (NH4)2C2O4 и 1 мл 25% NH4Cl. Доводят аммиаком pH раствора до 9 и переносят его в электролитическую ячейку, в которую предварительно помещают диск из нержавеющей стали, служащий мишенью для осаждения изотопов урана. Стакан обмывают 5 мл дистиллированной воды. Промывные воды также переносят в электролитическую ячейку.
7.8. Электролиз проводят при силе тока 2 А в течение 0,5 часа. В ходе электролиза контролируют pH раствора, поддерживая его на уровне pH = 9. За минуту до окончания электролиза в раствор добавляют 1 мл раствора аммиака. Разбирают ячейку, вынимают мишень (диск из нержавеющей стали), промывают ее этиловым спиртом и высушивают при комнатной температуре. Полученный препарат является счетным образцом для измерения активности изотопов урана на альфа-спектрометре.
7.9. Для дезактивации использованные диски кипятят в течение 1 часа в 0,5 H HNO3, промывают проточной водой, очищают содой и обрабатывают тонкой наждачной бумагой. Обработанные диски промывают дистиллированной водой, затем этиловым спиртом. Скорость счета от диска, измеренная на соответствующей аппаратуре, не должна превышать фоновых значений, в противном случае очистку повторяют.
7.10. Измерение активности 238U, 234U, 235U в приготовленном счетном образце и определение их удельной активности в исследуемой пробе выполняют в соответствии с главой 11 настоящих МР.
VIII. РАДИОХИМИЧЕСКОЕ ВЫДЕЛЕНИЕ 210Po и 210Bi(210Pb)
И ПРИГОТОВЛЕНИЕ СЧЕТНЫХ ОБРАЗЦОВ ДЛЯ ОПРЕДЕЛЕНИЯ
ИХ УДЕЛЬНОЙ АКТИВНОСТИ
8.1. Метод выделения полония основан на электрохимическом осаждении радионуклида на никелевом диске из солянокислых растворов. Этот метод называется электролитическим замещением или бестоковым электролизом. Определение 210Pb производят по дочернему 210Bi, который так же, как и 210Po, электрохимически осаждается на никелевом диске.
8.2. Анализ на содержание полония осложняется летучестью его соединений, особенно органических. Поэтому при определении содержания полония в неводных пробах рекомендуется использовать только мокрое озоление. При этом необходимо ограничивать температуру окончательного выпаривания пробы досуха с HNO3 и HCl до 120 - 150 °C.
8.3. Подготовленные согласно п. 3.3 пробы твердых продуктов взвешивают (вес записывают в рабочем журнале) и высушивают при комнатной температуре. Жидкие пробы концентрируют выпариванием с добавлением азотной кислоты до минимального объема.
8.4. Пробу помещают в стакан емкостью 600 - 800 мл и заливают 100 мл концентрированной HNO3, приливая ее небольшими порциями, чтобы избежать избыточного пенообразования. Пробу, залитую концентрированной HNO3, оставляют на ночь.
8.5. Затем продолжают проводить мокрое озоление пробы при нагревании до 120 - 150 °C. Раствор над пробой упаривают до минимального объема, снова приливают 50 мл концентрированной HNO3. Процесс повторяют несколько раз до полного сжигания органики.
8.6. Затем к пробе помимо концентрированной азотной кислоты добавляют по каплям 35% раствор перекиси водорода H2O2 (общий объем прибавляемого раствора составляет 4 - 8 мл) через каждые 10 - 15 минут и продолжают процесс окисления, следя за тем, чтобы он проходил спокойно. Процесс проводят до тех пор, пока не получают осветленный раствор, после чего его выпаривают до влажных солей.
8.7. Полученные нитраты переводят в хлориды, для этого к влажным солям небольшими порциями приливают около 50 мл концентрированной соляной кислоты (HCl) и вновь упаривают получаемый раствор до минимального объема. Описанный процесс повторяют 3 - 4 раза до исчезновения бурых паров окислов азота.
8.8. Последнюю порцию упаренного до минимального объема солянокислого раствора пробы выпаривают до сухих солей. По окончании выпаривания, в стакан приливают 75 мл 1 H HCl и кипятят несколько минут. Полученный раствор фильтруют через фильтр "синяя лента".
8.9. Ионы Fe3+ препятствуют выделению анализируемых радионуклидов на металлах, поэтому их переводят в Fe2+. Для восстановления железа до двухвалентного состояния в раствор вносят 100 - 200 мг сухой лимонной кислоты при перемешивании его до полного растворения внесенного реактива. Затем добавляют сухой реактив аскорбиновой кислоты до обесцвечивания раствора. Раствор доводят дистиллированной водой до 100 мл.
8.10. Электрохимическое осаждение 210Po и 210Bi проводят на диск из никелевой фольги, рекомендуется применять диски диаметром 34 мм и толщиной 0,6 мм. Диск закрепляют в тефлоновой кассете, которая обеспечивает выделение 210Po и 210Bi на одной стороне диска. Кассету с диском помещают в огнеупорный стеклянный стакан на 250 мл и приливают в него подготовленный раствор.
8.11. Стакан накрывают часовым стеклом. Содержимое стакана нагревают на кипящей водяной бане (90 °C) в течение 5 - 6 часов, периодически перемешивая раствор. Объем жидкости должен быть равным 100 мл в течение всего времени осаждения, поэтому необходимо добавлять к пробе дистиллированную воду по мере испарения раствора.
8.12. Затем вынимают диск из тефлоновой кассеты, промывают дистиллированной водой, этиловым спиртом и высушивают на воздухе. Записывают время окончания электрохимического осаждения - это время отделения 210Bi от 210Pb.
8.13. Проведение измерений счетного образца и определение удельной активности 210Po и 210Pb проводят согласно главе 12 настоящих МР.
8.14. Использованные диски перед повторным применением в зависимости от активности либо кипятят в 0,5 H (10%) растворе HNO3 до появления зеленоватого цвета в растворе, либо замачивают на некоторое время в азотной кислоте той же нормальности. Затем высушивают и полируют мелкой наждачной бумагой. Отполированные диски промывают водой с мылом, высушивают и протирают спиртом для обезжиривания.
IX. ПОРЯДОК ПРОВЕДЕНИЯ ИЗМЕРЕНИЙ АКТИВНОСТИ
СЧЕТНЫХ ОБРАЗЦОВ
9.1. Измерения активности счетных образцов с применением альфа-бета радиометра выполняют по схеме:
- проводят подготовку радиометра к работе согласно руководству по эксплуатации прибора;
- устанавливают в барабан радиометра чистую, пустую измерительную кювету и выполняют измерение фона в альфа- и/или бета-канале в течение 60 мин;
- измерения выполняют в соответствии с техническим описанием и инструкцией по эксплуатации радиометра;
- время измерения и количество накопленных импульсов (NФ) заносят в рабочий журнал;
- вычисляют фоновую скорость счета импульсов пф, имп/мин, по формуле:
 , (1)
, (1)
где: NФ - количество накопленных импульсов при измерении фона, имп;
tф - время измерения фона, мин.
- в случае, если nф превышает фоновое значение в бета- и/или альфа-канале, проводят дезактивацию барабана радиометра этиловым спиртом;
- устанавливают в барабан радиометра контрольный источник и выполняют измерение в течение 10 мин;
- время измерения tки, мин, и количество накопленных импульсов Nки, имп, заносят в рабочий журнал;
- вычисляют скорость счета импульсов от контрольного источника по альфа - и/или бета-каналу nки имп/мин, по формуле:
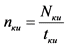 , (2)
, (2)
где: Nки - количество накопленных импульсов при измерении контрольного источника, имп;
tки - время измерения контрольного источника, мин.
- скорость счета импульсов от контрольного источника не должна отличаться от значения, указанного в свидетельстве о поверке, более чем на 5%. В том случае, если это условие не выполняется, выясняют причины, которые могли привести к изменению чувствительности радиометра. В случае необходимости проводят ремонт, наладку прибора. После ремонта проводят поверку прибора.
9.2. Определяют коэффициент связи радиометра для альфа- и бета-излучающих радионуклидов в соответствии с приложением 2 к настоящим МР.
9.3. Вычисляют скорость счета импульсов от счетного образца по бета- и альфа-каналу nсо, имп/мин, по формуле:
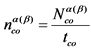 , (3)
, (3)
где: ![]() - количество накопленных импульсов при измерении счетного образца, имп;
- количество накопленных импульсов при измерении счетного образца, имп;
tсо - время измерения счетного образца, мин.
9.4. Химический выход для каждого из радионуклидов, измерения активности которых проводят с применением альфа-бета радиометра, вычисляют по формуле:
 , отн. ед. (4)
, отн. ед. (4)
где: ![]() - химический выход изотопа, отн. ед.;
- химический выход изотопа, отн. ед.;
m1 - количество носителя, определенного на выходе, мг;
m2 - количество носителя, добавленного в пробу, мг.
9.5. Измерения активности счетных образцов с применением полупроводникового альфа-спектрометра выполняют согласно требованиям руководства по эксплуатации прибора. Для определения эффективности регистрации альфа-излучения альфа-спектрометром проводят измерение контрольного источника (счетного образца) с известной активностью 238U. Эффективность регистрации альфа-излучения альфа-спектрометром вычисляют по формуле:
 , Бк/(имп/с) (5)
, Бк/(имп/с) (5)
где: AU-238 - активность контрольного источника (счетного образца), Бк;
tсо - время измерения счетного образца, с;
Sпика - площадь пика при измерении контрольного источника (счетного образца), имп.
Эффективность регистрации альфа-излучения определяют отдельно для каждой камеры альфа-спектрометра в которой проводят измерения счетных образцов подготовленных из анализируемых проб и для каждой полки, на которую счетные образцы помещают для измерения их активности.
9.6. Химический выход для каждого из изотопов урана, измерение активности которых проводят с применением альфа-спектрометра, вычисляют по формуле:
 , отн. ед. (6)
, отн. ед. (6)
где: ![]() - химический выход изотопа, отн. ед.;
- химический выход изотопа, отн. ед.;
232U1 - активность изотопного индикатора 232U, определенного на выходе, Бк;
232U2 - активность изотопного индикатора 232U, добавленного в пробу, Бк.
X. ОПРЕДЕЛЕНИЕ УДЕЛЬНОЙ АКТИВНОСТИ
228Ac(228Ra), 226Ra и 224Ra
10.1. После вычисления скорости счета импульсов от счетного образца La2O3(228AC) по бета-каналу согласно пп. 9.1 - 9.3, определяют удельную активность 228Ac(228Ra) в пробе по формуле:
 , Бк/кг, (7)
, Бк/кг, (7)
где: n - скорость счета импульсов от счетного образца за вычетом фона, имп/мин;
KAc-228 - коэффициент связи для 228Ac, Бк/(имп/мин) для измеряемой навески счетного образца, определенный согласно приложению 2 к настоящим МР;
![]() - постоянная распада 228Ac;
- постоянная распада 228Ac;
t - время, прошедшее от момента осаждения гидроокиси лантана (отделение 228Ac от 228Ra) до момента начала измерения скорости счета, час;
t1 - время накопления 228Ac в растворе 228Ra, час (при условии t1 < 40 часов);
m - масса пробы, кг;
![]() - химический выход лантана (п. 9.4);
- химический выход лантана (п. 9.4);
![]() - химический выход бария (п. 9.4).
- химический выход бария (п. 9.4).
Величины поправок на распад 228Ac (T1/2 = 6,13 часа) в зависимости от времени приведены в таблице 2.
Таблица 2
Поправка на распад 228Ac 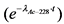 в зависимости от времени
в зависимости от времени
|
Время распада 228Ac, часы (t) |
|
Время распада 228Ac, часы (t) |
|
|
1 |
0,90 |
3,5 |
0,67 |
|
1,5 |
0,84 |
4 |
0,64 |
|
2 |
0,79 |
4,5 |
0,60 |
|
2,5 |
0,76 |
5 |
0,57 |
|
3 |
0,71 |
6 |
0,50 |
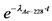
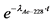
10.2. В течение первого месяца к испускаемой 226Ra альфа-частице добавляются еще три альфа-частицы, испускаемые 222Rn, 218Po и 214Po. Вследствие этого в данный период изменение альфа-активности 226Ra можно описать формулой:
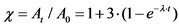 , (8)
, (8)
где: ![]() - поправочный коэффициент, учитывающий изменение альфа-активности 226Ra в первый месяц после его отделения от продуктов распада;
- поправочный коэффициент, учитывающий изменение альфа-активности 226Ra в первый месяц после его отделения от продуктов распада;
At - активность 226Ra в момент времени t после отделения его от продуктов распада, Бк/кг;
A0 - активность 226Ra в момент его выделения, когда t = 0, Бк/кг;
t - время от момента отделения 226Ra от продуктов его распада до измерения активности счетного образца, сутки;
![]() - постоянная распада 222Rn.
- постоянная распада 222Rn.
Соответствующие величины, характеризующие изменение альфа-активности 226Ra в течение первого месяца после выделения радионуклида, представлены в таблице 3.
Таблица 3
Изменение альфа-активности 226Ra в первый месяц
после выделения радионуклида 
|
t, сутки |
|
t, сутки |
|
|
1 |
1,5 |
14 |
3,76 |
|
2 |
1,92 |
15 |
3,81 |
|
3 |
2,26 |
17 |
3,86 |
|
4 |
2,45 |
18 |
3,89 |
|
5 |
2,80 |
20 |
3,92 |
|
6 |
2,98 |
21 |
3,93 |
|
7 |
3,15 |
22 |
3,95 |
|
8 |
3,28 |
28 |
3,98 |
|
10 |
3,52 |
30 |
3,99 |
|
11 |
3,60 |
36 |
4,00 |
10.3. Для определения активности 226Ra счетный образец (п. 6.12), измеряют через 4 часа, и через 1, 2 и 3 суток после прокаливания (можно провести измерения через иное количество суток при условии проведения не менее трех измерений в течение 11 суток после прокаливания) и делят результаты измерения на соответствующую времени измерения величину ![]() из таблицы 3.
из таблицы 3.
10.4. Совпадение (в пределах погрешности измерения) величин свидетельствует о наличии в образце преимущественно 226Ra и практическом отсутствии 224Ra, что позволяет определить активность 226Ra в период от 4 часов до 2 - 3 суток после отделения 226Ra от 222Rn. В этом случае вычисляют скорость счета импульсов от счетного образца (BaSO4) по альфа-каналу через 4 часа после прокаливания, а также через 1, 2 и 3 суток. Измеренные скорости счета импульсов, полученные через 1, 2 и 3 суток после прокаливания (т.е. после отделения 226Ra от 222Rn), делят на соответствующие времени измерения поправки из таблицы 3, рассчитывают среднее значение из четырех полученных величин (n1) и определяют удельную активность 226Ra по формуле:
 , Бк/кг, (9)
, Бк/кг, (9)
где: P - масса внесенного BaCl2, мг (в расчете на BaSO4);
n1 - средняя скорость счета импульсов от счетного образца через 4 часа, а также (с учетом внесения поправок из таблицы 3) через 1, 2 и 3 суток после прокаливания BaSO4(226Ra), имп/мин;
KRa-226 - коэффициент связи для 226Ra, (Бк/мг)/(имп/мин) для измеряемой навески счетного образца, определенный согласно приложению 2 к настоящим МР;
m - масса пробы, кг.
10.5. Если величины, полученные при измерении активности счетного образца через 1, 2 и 3 суток после отделения 226Ra от 222Rn, деленные на соответствующие значения ![]() из таблицы 3, не совпадают в пределах погрешности измерения, то это означает, что в пробе помимо 226Ra, в соизмеримых с ним количествах, присутствует 224Ra. В этом случае для идентификации 226Ra и 224Ra необходимо дополнительное измерение альфа-активности счетного образца через промежуток времени (t), достаточный для распада, находящегося в препарате 224Ra. Поскольку период полураспада 224Ra равен 3,64 суток, дополнительное измерение альфа-активности можно проводить, начиная с 11 суток после первого измерения. К этому времени 224Ra практически распадается, а нарастание измеренной активности счетного образца за счет накопления дочерних продуктов распада 226Ra учитывают, используя формулу (5).
из таблицы 3, не совпадают в пределах погрешности измерения, то это означает, что в пробе помимо 226Ra, в соизмеримых с ним количествах, присутствует 224Ra. В этом случае для идентификации 226Ra и 224Ra необходимо дополнительное измерение альфа-активности счетного образца через промежуток времени (t), достаточный для распада, находящегося в препарате 224Ra. Поскольку период полураспада 224Ra равен 3,64 суток, дополнительное измерение альфа-активности можно проводить, начиная с 11 суток после первого измерения. К этому времени 224Ra практически распадается, а нарастание измеренной активности счетного образца за счет накопления дочерних продуктов распада 226Ra учитывают, используя формулу (5).
10.6. Удельную активность 226Ra и 224Ra вычисляют, соответственно по формулам:
 , Бк/кг, (10)
, Бк/кг, (10)
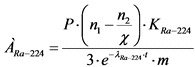 , Бк/кг, (11)
, Бк/кг, (11)
где: P - масса внесенного BaCl2, мг (в расчете на BaSO4);
n2 - скорость счета импульсов от счетного образца (за вычетом скорости счета фона) через t суток после первого измерения, имп/мин;
KRa-226 - и KRa-224 - коэффициенты связи для 226Ra и 224Ra, Бк/мг)/(имп/мин) для измеряемой навески счетного образца, определенные согласно приложению 2 к настоящим МР;
![]() - коэффициент, учитывающий регистрацию радиометром альфа-частиц, испускаемых дочерними продуктами распада 226Ra через t суток после первого измерения (таблица 3);
- коэффициент, учитывающий регистрацию радиометром альфа-частиц, испускаемых дочерними продуктами распада 226Ra через t суток после первого измерения (таблица 3);
m - масса пробы, кг;
n1 - скорость счета импульсов от счетного образца (за вычетом скорости счета фона) через 4 часа после прокаливания BaSO4(226Ra), имп/мин;
3 - коэффициент, учитывающий регистрацию радиометром альфа-частиц, испускаемых дочерними продуктами распада 224Ra;
![]() - постоянная распада 224Ra;
- постоянная распада 224Ra;
t - время, прошедшее от момента отделения 224Ra от 228Th (осаждение Ba(Ra)SO4) до момента первого измерения скорости счета счетного образца, сутки.
XI. ОПРЕДЕЛЕНИЕ УДЕЛЬНОЙ АКТИВНОСТИ 238U, 234U, 235U
11.1. Диск из нержавеющей стали, подготовленный согласно главе 7 настоящих МР, помещают на полку альфа-спектрометра, для которой определена эффективность регистрации альфа-излучения согласно п. 9.5 и проводят измерение активности альфа-излучающих изотопов урана согласно требованиям руководства по эксплуатации прибора.
11.2. Удельную активность каждого из альфа-излучающих изотопов урана (238U, 234U, 235U, 232U) в пробе определяют по формуле:
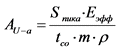 , Бк/кг, (12)
, Бк/кг, (12)
где: AU-a - альфа-излучающий изотоп урана (238U, 234U, 235U, 232U);
Sпика - площадь пика от соответствующего альфа-излучающего изотопа урана (238U, 234U, 235U, 232U), имп, регистрируемая альфа-спектрометром;
Eэфф - эффективности регистрации альфа-излучения альфа-спектрометром, Бк/(имп/с), определенный согласно п. 9.5;
tсо - время измерения счетного образца, с;
m - масса пробы, кг;
![]() - химический выход изотопов урана, определенный согласно п. 9.6.
- химический выход изотопов урана, определенный согласно п. 9.6.
XII. ОПРЕДЕЛЕНИЕ УДЕЛЬНОЙ АКТИВНОСТИ 210Po и 210Bi(210Pb)
12.1. Никелевый диск (счетный образец) после электролитического осаждения на него 210Po и 210Bi, помещают в альфа-бета радиометр и определяют скорость счета полученного образца по альфа- и бета-каналу согласно пп. 9.1 - 9.3.
12.2. Измерение активности счетного образца выполняют не ранее, чем через 10 - 12 часов после электрохимического выделения радионуклидов (например, на следующий день), поскольку кроме 210Po и 210Bi счетный образец может содержать короткоживущие альфа-активные (218Po, 214Po, 216Po, 212Po, 215Po, 211Po, 211Bi) и бета-активные (214Bi, 212Bi) радионуклиды. Выдержка счетного образца перед его измерением в течение 10 - 12 часов приводит к распаду этих короткоживущих радионуклидов, т.е. устраняет их мешающее влияние.
12.3. Удельную активность 210Po в пробе определяют по формуле:
 , Бк/кг (13)
, Бк/кг (13)
где: n - скорость счета импульсов от счетного образца за вычетом фона по альфа-каналу, имп/мин;
KPo-210 - коэффициент связи для 210Po, Бк/(имп/мин), полученный при использовании контрольного образца сравнения (приложение 2 к настоящим МР);
m - масса пробы, кг;
![]() - химический выход 210Po.
- химический выход 210Po.
12.4. Удельную активность 210Bi в пробе, которую принимают равной удельной активности 210Pb, определяют по формуле:
 , Бк/кг (14)
, Бк/кг (14)
где: n1 - скорость счета импульсов от счетного образца за вычетом фона по бета-каналу, имп/мин;
KBi-210 - коэффициент связи для 210Bi, Бк/(имп/мин), полученный при использовании контрольного образца сравнения (приложение 2 к настоящим МР);
m - масса пробы, кг;
![]() - химический выход 210Bi;
- химический выход 210Bi;
t - время от электрохимического выделения 210Bi до измерения его активности, часы;
![]() - постоянная распада 210Bi.
- постоянная распада 210Bi.
12.5. Химические выходы 210Po и 210Bi были определены экспериментально при разработке методики с использованием соответствующих стандартных растворов: 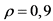 (для 210Po);
(для 210Po); 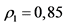 (для 210Bi);
(для 210Bi);
12.6. Поправки на распад 210Bi (T1/2 = 120 час) представлены в таблице 4.
Таблица 4
Поправка на распад 210Bi в зависимости от времени
|
Время распада 210Bi, часы (t) |
|
|
12 |
0,93 |
|
16 |
0,91 |
|
20 |
0,89 |
|
24 |
0,87 |
|
28 |
0,85 |
|
32 |
0,83 |
|
40 |
0,80 |
XIII. ОБРАБОТКА РЕЗУЛЬТАТОВ ИЗМЕРЕНИЙ
13.1. Результаты определения удельной активности радионуклидов в пробе представляют в форме:
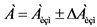 , Бк/кг (15)
, Бк/кг (15)
где: ![]() - результат измерения;
- результат измерения;
![]() - абсолютная погрешность измерения, численное значение которой рассчитывают по формуле:
- абсолютная погрешность измерения, численное значение которой рассчитывают по формуле:
 , Бк/кг (16)
, Бк/кг (16)
13.2. Значение ![]() принимают либо по таблице 1, либо рассчитывают, учитывая погрешности, возникающие на всех стадиях радиохимического анализа.
принимают либо по таблице 1, либо рассчитывают, учитывая погрешности, возникающие на всех стадиях радиохимического анализа.
Систематические погрешности складываются из:
- относительной погрешности определения химического выхода ![]() . В случае выделения изотопа без носителя включается относительная погрешность выхода изотопа, найденная в предварительных экспериментах;
. В случае выделения изотопа без носителя включается относительная погрешность выхода изотопа, найденная в предварительных экспериментах;
- относительной погрешности градуировки прибора, ![]() , определяемой как сумма паспортной погрешности определения активности эталона, погрешности при отборе аликвотной части раствора и погрешности измерения активности эталонного раствора.
, определяемой как сумма паспортной погрешности определения активности эталона, погрешности при отборе аликвотной части раствора и погрешности измерения активности эталонного раствора.
Среднеквадратическую погрешность измерения определяют по формуле:
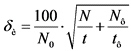 , % (17)
, % (17)
где: N0 - скорость счета от препарата за вычетом фона, имп/мин;
N - скорость счета от препарата с фоном, имп/мин;
t - время измерения препарата с фоном, мин;
Nф - скорость счета от фона, имп/мин;
tф - время измерения фона, мин.
Среднеквадратическая погрешность зависит от времени измерения и не должна превышать 30%.
Общую погрешность, ![]() , анализа рассчитывают по формуле:
, анализа рассчитывают по формуле:
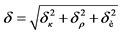 , % (18)
, % (18)
где: ![]() - относительная погрешность градуировки прибора, %;
- относительная погрешность градуировки прибора, %;
![]() - относительная погрешность определения химического выхода, %;
- относительная погрешность определения химического выхода, %;
![]() - среднеквадратическая погрешность измерения, %.
- среднеквадратическая погрешность измерения, %.
XIV. КОНТРОЛЬ КАЧЕСТВА РЕЗУЛЬТАТОВ ИЗМЕРЕНИЙ
14.1. Контроль качества результатов измерений при реализации методики в лаборатории предусматривает:
- контроль исполнителем процедуры выполнения измерений (на основе оценки погрешности при реализации отдельно взятой контрольной процедуры);
- контроль стабильности результатов измерения (на основе оценки погрешности при реализации всего анализа).
14.2. Контроль исполнителем процедуры выполнения измерений с использованием образцов для контроля (аттестованных смесей) проводят путем сравнения результата отдельно взятой контрольной процедуры (Кк) с нормативом контроля (К).
14.3. Результат контрольной процедуры (Кк) рассчитывают по формуле:
Кк= |Кх - Кс|, (19)
где: Кх - результат контрольного измерения активности радионуклида в образце для контроля;
Кс - аттестованное значение образца для контроля.
Качество контрольной процедуры признают удовлетворительным при выполнении условия:  , значения
, значения ![]() принимают по таблице 1 в соответствии с аттестованным значением образца для контроля.
принимают по таблице 1 в соответствии с аттестованным значением образца для контроля.
14.4. При невыполнении условия (19) эксперимент повторяют. При повторном невыполнении условия (19) выясняют причины, приводящие к неудовлетворительным результатам.
14.5. Периодичность контроля исполнителем процедуры выполнения измерений, а также реализуемые процедуры контроля стабильности результатов выполняемых измерений, регламентируют в Руководстве по качеству лаборатории.
Приложение 1
к МР 2.6.1/2.3.7.0216-20
ПРИГОТОВЛЕНИЕ РАБОЧИХ РАСТВОРОВ И РАСТВОРОВ НОСИТЕЛЕЙ.
ПОДГОТОВКА ИОНООБМЕННЫХ СМОЛ К АНАЛИЗУ,
РЕГЕНЕРАЦИЯ И ХРАНЕНИЕ
1. Приготовление кислот определенной нормальности:
Для выполнения операций по настоящими МР необходимо приготовить следующие кислоты: HCl - 0,5 H; 1 H; 2 H; 6 H; 8 H; HNO3 - 0,5 H.
Для приготовления рабочего раствора кислоты концентрированную кислоту разбавляют дистиллированной водой до нужной концентрации:
- Соляная кислота, 8 H раствор: 680 мл концентрированной HCl (35 - 38%) разбавляют дистиллированной водой до 1 л. Срок хранения не ограничен.
- Соляная кислота, 6 H раствор: 510 мл концентрированной HCl (35 - 38%) разбавляют дистиллированной водой до 1 л. Срок хранения не ограничен.
- Соляная кислота 2 H раствор: 170 мл концентрированной HCl (35 - 38%) разбавляют дистиллированной водой до 1 л. Срок хранения не ограничен.
- Соляная кислота 1 H раствор: 85 мл концентрированной HCl (35 - 38%) разбавляют дистиллированной водой до 1 л. Срок хранения не ограничен.
- Соляная кислота 0,5 H раствор: 42,5 мл концентрированной HCl (35 - 38%) разбавляют дистиллированной водой до 1 л. Срок хранения не ограничен.
- Азотная кислота 0,5 H раствор: 33,6 мл концентрированной азотной кислоты разбавляют дистиллированной водой до 1 л. Срок хранения не ограничен.
2. Приготовление рабочих растворов:
- Натрий углекислый, 5% раствор: 50 г соли растворяют при нагревании в 950 см3 дистиллированной воды. Срок хранения 1 год.
- Лимонная кислота, 1% раствор: 1 г кислоты растворяют в 99 мл дистиллированной воды. Срок хранения 1 год.
- Аммоний хлористый, 25% раствор: 25 г соли NH4Cl растворяют в 75 см3 дистиллированной воды. Срок хранения не ограничен.
- Аммоний щавелевокислый, насыщенный раствор: 12 г соли (NH4)2C2O4·H2O растворяют в 100 см3 дистиллированной воды. Срок хранения не ограничен.
- Серная кислота, 10% раствор: приготовляют по объему растворением 100 мл концентрированной H2SO4 в 900 мл дистиллированной воды. Срок хранения не ограничен.
- Аммоний углекислый, 15% раствор: 15 г (NH4)2CO3·H2O растворяют в 85 см3 дистиллированной воды. Срок хранения не ограничен.
- Аммоний углекислый, 10% раствор: 10 г (NH4)2CO3·H2O растворяют в 90 см3 дистиллированной воды. Срок хранения не ограничен.
- Аммоний углекислый, 2% раствор: 2 г (NH4)2CO3·H2O растворяют в 98 см3 дистиллированной воды. Срок хранения не ограничен.
- Аммиак водный безугольный, раствор: получают перегонкой NH4OH в аппарате, собранном из перегонной колбы, холодильника (ловушки) и приемной колбы. Для связывания CO2 в перегонную колбу добавляют Ba(OH)2 из расчета 5 - 10 г/л. Срок хранения 1 месяц.
3. Приготовление титрованного раствора бария.
Исходная соль для приготовления раствора - BaCl2·2H2O. Соединение, в виде которого выделяется носитель на последней стадии анализа - BaSO4. Объем приготовляемого раствора носителя - 100 мл. Заданный титр BaSO4 - 100 мг/мл, т.е. в 100 мл раствора должно содержаться 100 мг x 100 мл = 10 г BaSO4.
Молекулярная масса BaCl2·2H2O - 244,28.
Молекулярная масса BaSO4 - 233,4.
Расчет титра сводится к следующему:
|
244,28 |
-------- |
233,4 |
|
X (г) |
-------- |
10 (г) |
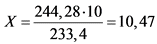 г
г
Для приготовления 100 мл раствора BaCl2 с титром BaSO4 ~ 100 мг/мл необходима навеска соли (BaCl2·2H2O) массой 10,47 г. Навеску помещают в стакан емкостью 150 мл, растворяют в дистиллированной воде, добавляют 5 - 10 мл 2 H HCl. Раствор переливают в мерную колбу емкостью 100 мл (если необходимо - фильтруют), доводят дистиллированной водой до метки.
Определение титра раствора проводят в условиях, аналогичных тем, которые используются на последнем этапе радиохимического анализа при выделении изотопов радия на BaSO4.
Для этого в пять стаканов емкостью 150 мл отбирают по 1 мл приготовленного раствора, добавляют 10 - 15 мл 2 H HCl и разбавляют дистиллированной водой до 100 мл. Нагревают раствор до кипения и осаждают BaSO4, приливая при непрерывном перемешивании 10 мл 10% H2SO4. После отстаивания в течение 30 - 40 минут осадки фильтруют через обеззоленный фильтр "синяя лента", тщательно перенося их со стенок стаканов горячей дистиллированной водой. Фильтры с осадками помещают в тигли, обугливают на электрической плитке, переносят в муфельную печь, прокаливают при 800 °C в течение 1 часа, после охлаждения взвешивают. Вычисляют среднее арифметическое значение массы осадка, которое считают точной концентрацией, т.е. титром бария 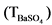 в растворе.
в растворе.
4. Приготовление титрованного раствора лантана.
Исходная соль для приготовления раствора - LaCl3·7H2O. Соединение, в виде которого выделяется носитель на последней стадии анализа - La2O3. Объем приготовляемого раствора носителя - 100 мл. Заданный титр La2O3 - 50 мг/мл, т.е. в 100 мл раствора должно содержаться 50 мг x 100 мл = 5 г La2O3.
Молекулярная масса LaCl3·7H2O - 371,38.
Молекулярная масса La2O3 - 325,82.
Для получения одной молекулы La2O3 необходимо две молекулы LaCl3·7H2O, молекулярная масса которых составляет 742,46.
Расчет титра сводится к следующему:
|
742,46 |
-------- |
325,82 |
|
X (г) |
-------- |
5 (г) |
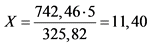 г
г
Для приготовления 100 мл раствора LaCl3 с титром La2O3 ~ 50 мг/мл необходима навеска соли (LaCl3·7H2O) массой 11,40 г. Навеску помещают в стакан емкостью 150 мл, растворяют при нагревании в 20 - 40 мл 2 H HCl. После охлаждения раствор переливают в мерную колбу емкостью 100 мл, доводят дистиллированной водой до метки.
Для определения титра приготовленного раствора используют приемы, указанные в методике выделения 228Ac на La(OH)3 на последнем этапе его радиохимического анализа. Для этого в пять стаканов емкостью 150 мл отбирают по 1 мл приготовленного раствора носителя, добавляют 10 мл 2 H HCl и разбавляют дистиллированной водой до 100 мл. Нагревают раствор до 80 °C и осаждают La(OH)3, добавляя раствор NH4OH до pH = 8 - 9. Осадки фильтруют через обеззоленный фильтр "белая лента", тщательно перенося их со стенок стаканов горячей дистиллированной водой. Фильтры с осадками помещают в тигли, обугливают на электрической плитке, переносят в муфельную печь, прокаливают при 800 °C в течение 1 часа, после охлаждения взвешивают. Вычисляют среднее арифметическое значение массы осадка, которое считают точной концентрацией, т.е. титром лантана  в растворе.
в растворе.
5. Приготовление титрованного раствора железа.
Исходная соль для приготовления раствора - FeCl3·6H2O. Соединение, в виде которого выделяется носитель на последней стадии анализа - Fe2O3. Объем приготовляемого раствора носителя - 100 мл. Заданный титр Fe2O3 - 20 мг/мл, т.е. в 100 мл раствора должно содержаться 20 мг x 100 мл = 2 г La2O3.
Молекулярная масса FeCl3·6H2O - 270,3.
Молекулярная масса Fe2O3 - 159,69.
Для получения одной молекулы Fe2O3 необходимо две молекулы FeCl3·6H2O, молекулярная масса которых составляет 540,6.
Расчет титра сводится к следующему:
|
540,6 |
-------- |
159,69 |
|
X (г) |
-------- |
2 (г) |
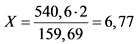 г
г
Для приготовления 100 мл раствора FeCl3 с титром Fe2O3 ~ 20 мг/мл необходима навеска соли (FeCl3·6H2O) массой 6,77 г. Навеску помещают в стакан емкостью 150 мл, добавляют 10 мл 2 H HCl и дистиллированную воду до 80 мл. Раствор переливают в мерную колбу емкостью 100 мл и доводят дистиллированной водой до метки.
Для определения титра раствора в пять стаканов емкостью 150 мл отбирают по 1 мл приготовленного раствора носителя, добавляют 10 мл 2 H HCl и разбавляют дистиллированной водой до 100 мл. Нагревают раствор до 80 °C и осаждают Fe(OH)3, добавляя раствор NH4OH до pH = 8 - 9. Осадки фильтруют через обеззоленный фильтр "белая лента", тщательно перенося их со стенок стаканов горячей дистиллированной водой. Фильтры с осадками помещают в тигли, обугливают на электрической плитке, переносят в муфельную печь, прокаливают при 800 °C в течение 1 часа, после охлаждения взвешивают. Вычисляют среднее арифметическое значение массы осадка, которое считают точной концентрацией, т.е. титром железа 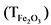 в растворе.
в растворе.
6. Приготовление титрованного раствора иттрия.
Исходная соль для приготовления раствора - YCl3·6H2O. Соединение, в виде которого выделяется носитель на последней стадии анализа - Y2O3. Объем приготовляемого раствора носителя - 100 мл. Заданный титр Y2O3 - 60 мг/мл, т.е. в 100 мл раствора должно содержаться 60 мг x 100 мл = 6 г Y2O3.
Молекулярная масса YCl3·6H2O - 303,4.
Молекулярная масса Y2O3 - 225,8.
Для получения одной молекулы Y2O3 необходимо две молекулы YCl3·6H2O, молекулярная масса которых составляет 606,8.
Расчет титра сводится к следующему:
|
606,8 |
-------- |
225,8 |
|
X (г) |
-------- |
6 (г) |
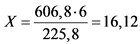 г
г
Для приготовления 100 мл раствора YCl3 с титром Y2O3 ~ 60 мг/мл необходима навеска соли (YCl3·6H2O) массой 16,12 г. Навеску помещают в стакан емкостью 150 мл, добавляют 15 - 20 мл 2 H HCl, нагревают до растворения осадка, разбавляют дистиллированной водой до 70 мл, охлаждают. Раствор переливают в мерную колбу емкостью 100 мл и доводят дистиллированной водой до метки.
Для определения титра раствора в пять стаканов емкостью 150 мл отбирают по 1 мл приготовленного раствора носителя, добавляют 10 - 15 мл 2 H HCl и дистиллированную воду до 60 - 70 мл. После нагревания до 70 °C добавляют 10 мл насыщенного раствора H2C2O4 и раствором и NH4OH доводят до pH ~ 1,5. После отстаивания (около 30 минут) осадки фильтруют через обеззоленный фильтр "синяя лента", промывают дистиллированной водой с добавлением H2C2O4. Фильтры с осадками помещают в тигли, обугливают на электрической плитке, переносят в муфельную печь, прокаливают при 800 °C в течение 1 часа, после охлаждения взвешивают. Вычисляют среднее арифметическое значение массы осадка, которое считают точной концентрацией, т.е. титром иттрия ![]() в растворе.
в растворе.
7. Подготовка ионообменных смол к анализу, регенерация и хранение.
Для удовлетворительного хроматографического разделения ионов следует пользоваться только мелкозернистыми ионитами. Размер зерен смол, используемых для разделения урана и тория - (0,125 ![]() 0,250) мм.
0,250) мм.
Подготовленной таким образом фракции ионита нужно дать набухнуть, причем в процессе набухания следует сливать верхнюю жидкость для удаления из смолы частиц почти коллоидного размера. Для этого в мерном цилиндре приготавливают взвесь смолы в десятикратном объеме воды и оставляют ее приблизительно на полчаса. Образовавшуюся над смолой облакообразную взвесь сливают, добавляют в цилиндр порцию воды и повторяют весь процесс до тех пор, пока слой жидкости над смолой не станет чистым. В течение всего этого процесса смола впитывает воду и набухает. Далее проводят подготовительный цикл, при котором ионит переводят в Cl--форму: смолы обрабатывают 1 - 2 H раствором HCl, дистиллированной водой, 0,5 - 1 H раствором NaOH и снова дистиллированной водой.
Для переведения смолы в Cl--форму ее обрабатывают 1 H раствором HCl и промывают дистиллированной водой. Каждый раз смолу нужно промывать дистиллированной водой до нейтральной реакции. Время обработки смолы каждым реактивом - несколько часов, при переведении в Cl--форму смолу, залитую 1 H раствором HCl, лучше оставить на ночь. По окончании анализа смолу переносят из колонок в стакан, промывают дистиллированной водой, сливают верхнюю жидкость и проводят полный подготовительный цикл.
После регенерации смолу тщательно промывают дистиллированной водой и хранят в закрытых сосудах под слоем дистиллированной воды. Смолу, которая хранилась в течение года или дольше, перед использованием рекомендуется вновь регенерировать.
Приложение 2
к МР 2.6.1/2.3.7.0216-20
ПРИГОТОВЛЕНИЕ КАЛИБРОВОЧНЫХ ОБРАЗЦОВ
И ГРАДУИРОВКА АЛЬФА-БЕТА РАДИОМЕТРА
1. Градуировка альфа-бета радиометра для измерения активности альфа-излучающих радионуклидов (226Ra, 224Ra).
Коэффициенты связи между активностью и скоростью счета импульсов от счетного образца альфа-излучающих радионуклидов для толстослойных препаратов, имеющих поверхностную плотность более 10 мг/см2, (Бк/мг)/(имп/мин), определяют с использованием эталонного раствора 226Ra с объемной активностью 3 - 25 Бк/см3.
Для приготовления калибровочных образцов используют аликвоты эталонного раствора (готовят 10 - 15 образцов) 226Ra одинаковой активности. К отобранным аликвотам эталонного раствора 226Ra одинакового объема, перенесенным в термостойкие химические стаканы емкостью 100 мл приливают по 20 - 30 мл 2 H HCl, вносят раствор носителя 226Ra - хлористый барий BaCl2 (от 10 - 15 до 200 мг в расчете на BaSO4) и доводят объем раствора до 100 мл дистиллированной водой. Приготовленные растворы кипятят 10 - 15 минут на электрической плитке, добавляют при непрерывном перемешивании 15 мл 10% H2SO4, кипятят 10 - 15 мин., после чего осадку (Ba(Ra)SO4) дают отстояться в течение 1 часа. Осадок отфильтровывают через обеззоленный фильтр "синяя лента" (масса золы одного фильтра - 0,0004 г) и несколько раз промывают дистиллированной водой. Промытый осадок сульфата бария Ba(Ra)SO4 высушивают, прокаливают в муфельной печи при температуре 800 °C в течение 1 часа, взвешивают, растирают в тигле стеклянной палочкой до состояния "пудры" и переносят шпателем на предварительно взвешенную подложку для измерения активности 226Ra. Смачивают перенесенный на подложку осадок несколькими каплями этилового спирта, с помощью стеклянной палочки равномерно распределяют и уплотняют на подложке, затем высушивают. Высушенный осадок взвешивают (вместе с подложкой) для определения массы полученного образца.
Через 3 - 4 часа после прокаливания проводят измерение полученных образцов. Вычисляют скорость счета импульсов от каждого приготовленного счетного образца по альфа-каналу. Рассчитывают коэффициент связи (KRa-226) между активностью и скоростью счета импульсов от излучения 226Ra в толстослойном образце по формуле:
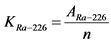 , (Бк/мг)/(имп/мин), (2.1)
, (Бк/мг)/(имп/мин), (2.1)
где: ARa-226 - массовая активность толстослойного градуировочного образца (BaSO4), приготовленного из отобранной аликвоты эталонного раствора, Бк/мг;
n - средняя скорость счета от серии приготовленных толстослойных образцов BaSO4, имп/мин.
Полученный коэффициент связи между активностью и скоростью счета импульсов от излучения 226Ra в счетных образцах используется только для расчетов активности альфа-излучения толстослойных образцов, измеренных в идентичных условиях:
- калибровочные и счетные образцы, приготовленные для измерения, должны иметь одинаковые размеры;
- геометрия измерения указанных образцов должна быть одинаковой.
При изменении условий измерения, например, при использовании подложек иной площади, процедуру калибровки повторяют, применяя для приготовления счетных образцов соответствующие подложки.
Приготовленный эталон можно использовать для проверки коэффициента связи в течение длительного времени. По прошествии 36 суток от его первичного измерения значение коэффициента связи рассчитывают по формуле:
 , (Бк/мг)/(имп/мин) (2.2)
, (Бк/мг)/(имп/мин) (2.2)
Измерения приготовленных калибровочных образцов (разной массы) повторяют в течение нескольких дней и рассчитывают коэффициенты связи. При расчете коэффициентов связи используют поправочные коэффициенты, характеризующие изменение альфа-активности 226Ra в течение первого месяца после выделения радионуклида, приведенные в таблице 3. На основании усредненных результатов измерения калибровочных образцов 226Ra (разных масс) строят график зависимости коэффициента связи от массы образца на подложке.
Расчет коэффициентов связи для 224Ra проводят по коэффициентам связи, полученным для 226Ra, учитывая несоответствие их энергетических спектров по формуле:
KRa-224 = 0,77 - KRa-226, (Бк/мг)/(имп/мин) (2.3)
2. Градуировка альфа-бета радиометра для измерения активности 210Po и 210Bi(210Pb).
Градуировка альфа-бета радиометра для измерения тонкослойных препаратов 210Po и 210Bi(210Pb) производится с помощью контрольного образца сравнения, в качестве которого используют диск из нержавеющей стали, на который методом электролитического осаждения нанесены эталонные растворы указанных радионуклидов.
Измерение активности радионуклидов 210Po, 210Bi, 210Pb в контрольном образце сравнения проводится в соответствии с пп. 9.1 - 9.3.
Коэффициент связи (KPo-210) между альфа-активностью 210Po в контрольном образце сравнения и скоростью счета импульсов от альфа-излучения 210Po в этом образце Бк/(имп/мин) определяют по формуле:
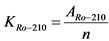 , Бк/(имп/мин) (2.4)
, Бк/(имп/мин) (2.4)
где: ARo-210 - активность 210Po в контрольном образце сравнения, Бк;
n - скорость счета импульсов за вычетом скорости счета фона, имп/мин.
Коэффициент связи (KBi-210) между бета-активностью 210Bi в контрольном образце сравнения и скоростью счета импульсов от бета-излучения 210Bi в этом образце определяют по формуле:
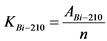 , Бк/(имп/мин) (2.5)
, Бк/(имп/мин) (2.5)
где: ABi-210 - активность 210Bi в контрольном образце сравнения, Бк;
n - скорость счета импульсов за вычетом скорости счета фона, имп/мин.
2. Градуировка альфа-бета радиометра для измерения активности 228Ra(228Ac).
Энергия бета-излучения 228Ac, находящегося в радиоактивном равновесии с 228Ra, и 234Pa, близка к энергии бета-излучения 90Y (234Pa - 2300 кэВ, 228Ac - 2089 кэВ, 90Y - 2274 кэВ), поэтому градуировку альфа-бета радиометра для измерения активности 228Ra(228Ac) проводят с применением калибровочных образцов 90Sr(90Y). Для приготовления калибровочных образцов 90Sr(90Y) используется эталонный раствор радионуклидов 90Sr(90Y) объемной активностью 3 - 25 Бк/см3. К отобранным аликвотам эталонного раствора одинакового объема, перенесенным в термостойкие химические стаканы (готовят 10 - 15 образцов), добавляют 20 - 30 мл 2 H HCl, вносят растворы носителей 90Y - хлористый иттрий (от 20 до 200 мг в расчете на Y2O3) и хлористый стронций (100 мг в расчете на SrSO4), разбавляют дистиллированной водой до 100 мл и кипятят в течение 20 - 30 минут. Из горячего раствора осаждают гидроокись иттрия добавлением безугольного аммиака до pH = 8 - 9, осадок отфильтровывают и промывают горячей дистиллированной водой. Время отделения 90Y от 90Sr записывают и учитывают при расчете активности (таблица 2.1). Осадок переносят в фарфоровый тигель и прокаливают в муфеле при температуре 800 - 900 °C до получения окиси иттрия Y2O3. Полученный осадок наносят равномерным слоем на предварительно взвешенную подложку, взвешивают осадок вместе с подложкой для определения массы полученного образца. Таким образом, готовят 10 - 15 образцов с разной массой для измерения активности 90Y. Вычисляют скорость счета импульсов от приготовленных счетных образцов по бета-каналу.
Рассчитывают коэффициент связи (KRa-228) между активностью и скоростью счета импульсов от излучения 90Sr(90Y) в толстослойных образцах разной массы (для каждой из масс) по формуле:
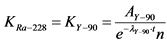 , Бк/(имп/мин) (2.6)
, Бк/(имп/мин) (2.6)
где: AY-90 - активность толстослойного градуировочного образца Y2O3, приготовленного из отобранной аликвоты эталонного раствора;
![]() - постоянная распада 90Y;
- постоянная распада 90Y;
t - время, прошедшее от момента отделения 90Y от 90Sr до времени измерения активности образца, час;
п - скорость счета от приготовленного толстослойного образца Y2O3, имп/мин.
Величины поправок на распад 90Y (T1/2 = 64,1 часа) в зависимости от времени приведены в таблице 2.1.
Таблица 2.1
Поправка на распад 90Y 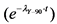 в зависимости от времени
в зависимости от времени
|
Время распада 90Y, часы (t) |
|
Время распада 90Y, часы (t) |
|
|
2 |
0,98 |
45 |
0,62 |
|
5 |
0,94 |
50 |
0,58 |
|
17 |
0,83 |
64 |
0,50 |
|
21 |
0,80 |
72 |
0,47 |
|
25 |
0,76 |
96 |
0,35 |
|
30 |
0,72 |
120 |
0,27 |
|
36 |
0,68 |
144 |
0,31 |
Строят график зависимости коэффициентов связи от массы счетного образца на подложке.