"Клинические рекомендации "Первичный иммунодефицит - X-сцепленный лимфопролиферативный синдром"
МИНИСТЕРСТВО ЗДРАВООХРАНЕНИЯ РОССИЙСКОЙ ФЕДЕРАЦИИ
КЛИНИЧЕСКИЕ РЕКОМЕНДАЦИИ
ПЕРВИЧНЫЙ ИММУНОДЕФИЦИТ - X-СЦЕПЛЕННЫЙ
ЛИМФОПРОЛИФЕРАТИВНЫЙ СИНДРОМ
МКБ 10: D82.3, D82.8, D84.8, D89.8
Год утверждения (частота пересмотра): 2018 (не реже 1 раза в 3 года)
ID: КР544/1
URL
Профессиональные ассоциации
- Национальное общество детских гематологов и онкологов Национальное общество экспертов по первичным иммунодефицитам Российская ассоциация аллергологов и клинических иммунологов
Ключевые слова
- Первичный иммунодефицит
- Гемофагоцитарный лимфогистиоцитоз
- Эпштейн-Барр-ассоциированное заболевание
- Воспалительное заболевание кишечника у детей
- Внутривенный иммуноглобулин
- Пренатальная диагностика
- Трансплантация гематопоэтических стволовых клеток
- X-сцепленный лимфопролиферативный синдром
Список сокращений
ВЗК - воспалительное заболевание кишечника
ГЛГ - гемофагоцитарный лимфогистиоцитоз
ДНК - дезоксирибонуклеиновая кислота
ЖКТ - желудочно-кишечный тракт
ЛДГ - лактатдегидрогеназы
НК - натуральные киллеры
НКТ - натуральные киллерные T-клетки
ПИД - Первичный иммунодефицит
ПЦР - полимеразной цепной реакции
ТГСК - трансплантация гематопоэтических стволовых клеток
УЗИ - Ультразвуковое исследование
рчГ-КСФ - гранулоцитарный колониестимулирующий фактор рекомбинантный
ФМН - фульминантный мононуклеоз
XЛП - X-сцепленный лимфопролиферативный синдром
ЦМВ - цитомегаловирус
ЭБВ - Вирус Эпштейна-Барр
ЭКГ - Электрокардиография
BIRC4 - baculoviral IAP repeat-containing protein 4
HHV VI - герпес-вируса 6 типа
HLA - human leukocyte antigen
IL - интерлейкин
IUIS - Международный союз иммунологических обществ
L18-MDP - L18-Muramyl dipeptide
MAIT - Mucosa-associated invariant T cells
MCP-1 - моноцитарный хемотаксический протеин-1
RICD - Restimulation-induced cell death
SLAM - Signaling lymphocyte activation molecule
TCR - T-клеточный рецепторов
TLR - toll-like receptors
TNF-alpha - фактор некроза опухоли альфа
Treg - регуляторные T-клетки
XIAP - X-linked inhibitor of apoptosis - X-сцепленный ингибитор апоптоза
XMEN - X-сцепленный иммунодефицит с дефектом магниевых каналов, ЭБВ-инфекцией и неоплазией
Термины и определения
Внутривенные иммуноглобулины - препараты, содержащие преимущественно нормальный человеческий IgG. Изготовляются из пулированной плазмы тысяч здоровых доноров, с применением специальных методов очистки и вирусинактивации.
Воспалительное заболевание кишечника - заболевание, характеризующееся хроническим воспалением желудочно-кишечного тракта. Включает в себя болезнь Крона и неспецифический язвенный колит.
Гемофагоцитарный лимфогистиоцитоз - жизнеугрожающее состояние, вызванное неконтролируемой активацией макрофагов и цитотоксических лимфоцитов в совокупности с гиперпродукцией воспалительных цитокинов (цитокиновый шторм).
Нейтропения - снижение уровня нейтрофилов в периферической крови менее 1,5 x 109/л (для детей первого года жизни - менее 1,0 x 109/л).
Полимеразная цепная реакция - метод молекулярной биологии, позволяющий амплифицировать (размножить) определенный участок ДНК
Секвенирование ДНК - определение ее нуклеотидной последовательности. В результате секвенирования получают описание первичной структуры линейной ДНК в виде последовательности нуклеотидов в текстовом виде.
Трансплантация гематопоэтических стволовых клеток - метод лечения некоторых наследственных и приобретенных гематологических, онкологических и иммунных заболеваний, основанный на замене собственного, патологического, кроветворения пациента на нормальное кроветворение донора.
X-сцепленный рецессивный тип наследования - наследование мутации генов, расположенных на X хромосоме. При этом лица женского пола как правило являются бессимптомными носителями, а заболеванием страдают лишь лица мужского пола.
1. Краткая информация
1.1 Определение
X-сцепленный лимфопролиферативный синдром (XЛП) - это комбинированный первичный иммунодефицит (ПИД), характеризующийся атипичной реакцией на инфекцию вирусом Эпштейна-Барр (ЭБВ), вследствие чего развивается гемофагоцитоз, дисгаммаглобулинемия, аутоиммунная патология и, в зависимости от типа, злокачественной лимфопролиферация [1].
1.2 Этиология и патогенез
На сегодняшний день охарактеризованы три гена, мутации которых ведут к проявлениям XЛП - SH2D1A, XIAP (он же BIRC4) и MAGT1. Подтипы XЛП объединены в одну группу в связи с наследованием по X-сцепленному типу и в связи со схожестью клинической картины, представленной в первую очередь предрасположенностью к ЭБВ-ассоциированной лимфопролиферации и к развитию гемофагоцитарного лимфогистиоцитоза (ГЛГ). Тем не менее, имеются некоторые различия в клинической картине и существенные - в патогенезе XЛП различных типов. Наиболее изученными являются XЛП 1 и 2 типа, вызываемые мутациями в генах SH2D1А и XIAP соответственно.
Патогенез X-сцепленного лимфопролиферативного синдрома 1 типа
Ген SH2D1A кодирует SLAM-ассоциированный белок SAP, где SLAM - signaling lymphocyte activation molecule - сигнальная молекула активации лимфоцитов, экспрессирующаяся в T-, НК (натуральные киллеры), НКТ (натуральные киллерные T-клетки) и, по некоторым источникам, в трансформированных B-клетоках [2]. SAP является адаптерной молекулой в пути передачи сигнала от SLAM-подобных рецепторов внутрь клетки [3].
Мутация в гене SH2D1A приводит к следующим иммунологическим дефектам: нарушению эффекторных функций НК-клеток, CD8+ и CD4+ T-клеток, резкому снижению/отсутствию НКТ-клеток, гуморальным нарушениям в виде дисгаммаглобулинемии и уменьшению CD27+ B-клеток памяти.
В норме стимуляция SLAM-рецепторов в НК-клетках приводит к повышению концентрации  и в целом к активации НК-клеток. Соответственно у пациентов с XЛП1 НК-опосредованная цитотоксичность снижена [4]. Страдает у них и CD8+-опосредованная цитотоксичность, однако преимущественно на антигены, презентируемые B-клетками, что может частично объяснять предрасположенность именно к ЭБВ-инфекции и B-клеточным лимфомам [5]. Вклад в предрасположенность к ЭБВ-инфекции и лимфомам также вносит характерное для XЛП1 нарушение созревания НКТ-клеток, выражающееся в их снижении или полном отсутствии [6].
и в целом к активации НК-клеток. Соответственно у пациентов с XЛП1 НК-опосредованная цитотоксичность снижена [4]. Страдает у них и CD8+-опосредованная цитотоксичность, однако преимущественно на антигены, презентируемые B-клетками, что может частично объяснять предрасположенность именно к ЭБВ-инфекции и B-клеточным лимфомам [5]. Вклад в предрасположенность к ЭБВ-инфекции и лимфомам также вносит характерное для XЛП1 нарушение созревания НКТ-клеток, выражающееся в их снижении или полном отсутствии [6].
Имеющиеся при XЛП1 дефекты гуморального иммунитета обусловлены тем, что в отсутствии SAP снижается способность фолликулярных T-клеток формировать стабильные контакты с B-клетками. В результате нарушается дифференцировка наивных B-клеток в долгоживущие, высокоаффинные клетки памяти и плазматические клетки. Это объясняет малочисленность B-клеток памяти у пациентов с XЛП1 и гипогаммаглобулинемию и/или нарушение антиген-специфичного антительного ответа. Кроме того, это соответствует картине, описанной D. Purtilo и соавт. [7] при открытии заболевания - отсутствие герминативных центров в лимфоидной ткани большинства пациентов.
Наконец, исследования A.Snow и соавт. [8] показали вовлеченность SLAM-пути в процесс смерти, индуцируемой избыточной стимуляцией (restimulation-induced cell death - RICD). Запрограммированная клеточная гибель необходима для элиминации активированных T-клеток во время иммунного ответа, что ограничивает иммунный ответ и предотвращает лимфопролиферацию и избыточное T-опосредованное повреждение тканей. Вероятно, дефект пути RICD отвечает за значительное увеличение количества CD8+ T-клеток в ответ на ЭБВ-инфекцию у пациентов с XЛП1 и развитие ГЛГ.
Патогенез X-сцепленного лимфопролиферативного синдрома 2 типа
Ген XIAP, также известный как BIRC4 (baculoviral IAP repeat-containing protein 4), - кодирует цитоплазматический белок XIAP. Данный белок экспрессируется повсеместно, более выражена его экспрессия в гематопоэтических клетках, где она сопоставима в различных субпопуляциях [9].
XIAP является физиологическим ингибитором каспаз 3, 7, 9 и, следовательно, ингибитором клеточной гибели. У пациентов с XЛП2 лимфоциты подвержены избыточному апоптозу, что может объяснить отсутствие лимфом у пациентов с XЛП2. При этом, несмотря на повышенную склонность к апоптозу, снижение количества T- и B-лимфоцитов у пациентов с XЛП2, как правило, не развивается, за исключением снижения количества НКТ-клеток. Предполагается, что они более чувствительны к апоптозу, индуцированному активацией, а, следовательно, более зависимы от белка XIAP [10].
Помимо антиапоптотической функции XIAP опосредует сигнальный путь через NOD1/2 и Dectin-1 рецепторы. Данные рецепторы экспрессируются в эпителиальных клетках, миелоидных клетках и клетках Панета кишечника и представляют собой соответственно внутриклеточные и трансмембранные паттерн-распознающие рецепторы, улавливающие продукты деградации бактерий, грибов и некоторых вирусов. В норме активация этих рецепторов в моноцитах приводит к увеличению выработки цитокинов и хемокинов, важных для клиренса патогенов - IL (интерлейкин)-8, MCP-1 (моноцитарный хемотаксический протеин-1), IL-10. IL-8 и MCP-1 важны для миграции нейтрофилов, в то время как IL-10 - для сохранения гомеостаза кишечника. Не последнюю роль в поддержании баланса провоспалительных и противовоспалительных факторов и обеспечения защиты от чужеродных патогенов в кишечнике играют регуляторные T-клетки (Treg), Mucosa-associated invariant T cells (MAIT) and НКТ клетки, которые в отсутствии XIAP становятся подвержены апоптозу. Таким образом, нарушение цитокиновой продукции и избыточный апоптоз регуляторов гомеостаза кишечника ведут к развитию воспалительного заболевания кишечника (ВЗК) у пациентов с XЛП2 [11].
Среди других цитокинов, выработка которых снижена при дефиците XIAP, важно выделить фактор некроза опухоли альфа (TNF-alpha). Выработка цитокина моноцитами пациента в ответ на их стимуляцию лигандом NOD-рецептора L18-MDP (Muramyl dipeptide) снижена по сравнению с моноцитами здорового контроля - тест, который используется для диагностики XЛП2 [12].
В отличие от других форм семейных ГЛГ, при которых нарушена цитотоксическая функция лимфоцитов, при дефиците XIAP цитотоксичность T- и НК-клеток в норме. Будучи ингибитором апоптоза XIAP обеспечивает выживание и пролиферацию активированных T-клеток в случае инфекции, что способствует элиминации инфицированных клеток. Отсутствие белка ведет к персистенции инфицированных клеток. Более того, XIAP ингибирует TLR (toll-like receptors)- и TNF-опосредованное образование инфламмасомы в дендритных клетках и макрофагах, отсутствие же белка ведет к чрезмерной выработке IL-1, IL-18. В совокупности персистенция инфекции и системное воспаление провоцируют развитие ГЛГ [13].
Патогенез X-сцепленного лимфопролиферативного синдрома 3 типа
XЛП3 был назван XMEN - X-сцепленный иммунодефицит с дефектом магниевых каналов, ЭБВ-инфекцией и неоплазией. К его наиболее постоянным признакам относятся CD4+-лимфопения, спленомегалия, персистенция ЭБВ и высокая предрасположенность к развитию ЭБВ-ассоциированной лимфомы [14].
Ген MAGT1 экспрессируется во всех клетках, однако в некоторых сильнее, например, в гемопоэтических. Потеря функции гена MAGT1 в T-клетках ведет к нарушению быстрого потока свободных ионов магния в клетку и, как следствие, к нарушению антигенной стимуляции T-клеточных рецепторов (TCR). В НК-клетках и цитотоксических (CD8+) T-лимфоцитах магниевые каналы обеспечивают базальное содержание свободного магния, необходимое для поддержания экспрессии некоторых активирующих рецепторов. Соответственно, при отсутствии свободного магния цитотоксическая функция T-клеток страдает. Клинически увеличивается предрасположенность к развитию опухолей и инфекций, в частности ЭБВ-инфекции. Поскольку дефект активации T-клеток при синдроме XMEN не столь значителен и может быть компенсирован более длительной и сильной стимуляцией, у таких пациентов не наблюдается тяжелых угрожающих жизни инфекций, как при других ПИД. В отличие от других типов XЛП для пациентов с синдромом XMEN не характерно развитие фульминантного мононуклеоза (ФМН), редко наблюдается ГЛГ и количество НКТ-клеток у них в норме [15].
1.3 Эпидемиология
Частота встречаемости XЛП составляет 1 - 3 на 1 миллион рожденных мальчиков, заболеванием страдают лица мужского пола (за очень редким исключением) [6, 16 - 18].
1.4 Кодирование по МКБ-10
D82.3 Иммунодефицит вследствие наследственного дефекта, вызванного вирусом Эпштейна-Барр
D84.8 Другие уточненные иммунодефицитные нарушения (код также используется в КР по врожденной нейтропении)
D89.8 Другие уточненные нарушения с вовлечением иммунного механизма, не классифицированные в других рубриках
D82.8 Иммунодефицит, связанный с другими уточненными значительными дефектами
1.5 Классификация
По данным классификации ПИД 2017 г., утвержденной Международным союзом иммунологических обществ (IUIS), XЛП относится к группе иммунодефицитов с иммунной дисрегуляцией, подгруппе иммунодефицитов с предрасположенностью к ЭБВ-инфекции и лимфопролиферации. Исторически заболевание классифицируется как XЛП1, XЛП2, XЛП3, в зависимости от типа генетического дефекта - мутации генов SH2D1A, XIAP и MAGT1, соответственно [19].
1.6 Клиническая картина
Общим для XЛП является предрасположенность к ЭБВ-инфекции с развитием фульминантного мононуклеоза, который фактически представляет собой ГЛГ. Для XЛП2 также описаны случаи ГЛГ, вызванного персистенцией цитомегаловируса (ЦМВ) или герпес-вируса 6 типа (HHV VI). Кроме того, гемофагоцитоз у пациентов с XЛП2, как правило, носит менее агрессивный характер.
Еще одним часто встречающимся проявлением XЛП у детей и взрослых является развитие лимфом, имеющих в большинстве своем B-клеточную природу и локализующихся в илеоцекальной области. При этом развитие злокачественной лимфопролиферации возможно, как у ЭБВ-негативных, так и у ЭБВ-позитивных пациентов. Исключение составляет XЛП2, при котором к настоящему моменту не зафиксировано ни одного случая лимфомы [20].
Другим существенным отличием XЛП2 является развитие воспалительного заболевания кишечника, напоминающего гистологически болезнь Крона [21]. Дефицит XIAP относится к группе моногенных заболеваний с поражением желудочно-кишечного тракта, основной характеристикой которых является развитие ВЗК-подобного колита с ранней манифестацией.
К редким проявлениям XЛП1 относятся апластическая анемия, лимфоидный васкулит, лимфоидный гранулематоз легких [22].
К редким проявлениям XЛП2 относятся аутовоспалительные проявления, такие как артрит, увеит, узловатая эритема [10].
Клиническая картина может быть представлена одним или комбинацией приведенных выше состояний, при этом проявления у представителей одной и той же семьи могут различаться.
2. Диагностика
Детям и взрослым пациентам с подозрением на X-сцепленный лимфопролиферативный синдромом проводится дифференциальный диагноз в первую очередь проводить с:
- Другими формами семейного ГЛГ;
- Лимфомами при других ПИД;
- X-сцепленной агаммаглобулинемией;
- Другими ПИД, осложненными течением ВЗК.
2.1 Жалобы и анамнез
- У всех пациентов (или родителей) с подозрением на X-сцепленный лимфопролиферативный синдром рекомендуется проводить сбор анамнеза и жалоб для верификации диагноза [20 - 23].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 3)
Комментарии: Пациенты и их родители могут предъявлять жалобы на появившийся с раннего возраста жидкий стул, кровь в стуле, наличие увеличенных лимфоузлов, эпизоды лихорадки без явного очага инфекции и ответа на антибактериальную терапию, которые могут сопровождаться увеличением печени/селезенки. Следует уточнить особенности физического развития пациента, прибавку в весе, сроки возникновения, частоту и тяжесть кишечных проявлений, а также болел ли пациент инфекционным мононуклеозом и насколько тяжело протекало заболевание. При сборе семейного анамнеза обращать внимание на случаи ранних смертей мальчиков с признаками гемофагоцитоза, лимфопролиферации, а также случаи тяжелого колита у лиц мужского пола. У пациентов XЛП нередко отмечаются "немотивированные" субфебрилитеты и лихорадка без явного очага инфекции (как проявление гемофагоцитоза). Кроме того, эпизоды лихорадки и\или субфебрилитета с потами и потерей массы тела могут быть признаками злокачественного заболевания (например, так называемые "B" симптомы при лимфоме).
2.2 Физикальное обследование
- Всем пациентам с подозрением на X-сцепленный лимфопролиферативный синдром рекомендуется физикальный осмотр для верификации диагноза [9].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 3)
Комментарии: Пациенты с XЛП (особенно XЛП2 с симптомами колита) могут отставать в массо-ростовых показателях.
У детей и взрослых пациентов с XЛП 1 и 2 типа могут отмечаться пятнисто-папулезные сыпи, характерные для ГЛГ. При развитии цитопении, в том числе тромбоцитопении, отмечается геморрагическая сыпь.
При осмотре рекомендовано оценить размеры периферических лимфоузлов. Для пациентов с XЛП нередко характерна генерализованная лимфаденопатия или увеличение одной группы лимфоузлов в случае развития лимфомы.
Увеличение печени и селезенки - как проявление хронической ЭБВ-инфекции, ГЛГ или лимфомы.
2.3 Лабораторная диагностика
- Всем пациентам с подозрением на X-сцепленный лимфопролиферативный синдром рекомендуется исследование общего анализа крови для диагностики анемии и нейтропении [3, 7].
Уровень убедительности рекомендаций A (уровень достоверности доказательств - 1)
Комментарии: У пациентов с XЛП может выявляться лимфопения (менее 1000 клеток в мкл). При течении ГЛГ отмечается в первую очередь тромбоцитопения, в дальнейшем - анемия и нейтропения. При ВЗК возможно развитие тромбоцитоза как отражение хронического воспалительного процесса
- Всем пациентам с подозрением на X-сцепленный лимфопролиферативный синдром рекомендуется исследование биохимического анализа крови: мочевины, креатинина, фракций билирубина, аспартатаминотрансферазы (АсАт), аланинаминотрансферазы (АлАт), альбумина, лактатдегидрогеназы (ЛДГ), щелочной фосфатазы (ЩВ), глюкозы, триглицеридов, ферритина, фибриногена, C-реактивного белка для диагностики сопутствующих заболеваний и верификации диагноза [2, 3, 5, 7].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 3)
Комментарии: Повышение ферритина, триглицеридов, снижение фибриногена, гипоальбуминемия относятся к критериям ГЛГ. Повышение ЛДГ особенно характерно для лимфом.
- Всем пациентам с подозрением на X-сцепленный лимфопролиферативный синдром рекомендуется исследование концентрации иммуноглобулинов сыворотки IgG, IgA, IgM для верификации диагноза [9, 24].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 3)
Комментарии: В большинстве случаев у пациентов с XЛП имеются те или иные нарушения концентрации иммуноглобулинов - снижение всех классов иммуноглобулинов, или повышение одних классов и снижение других. При ВЗК в большинстве случаев отмечается повышение IgA, что отражает вовлеченность слизистых в воспалительный процесс
- Всем пациентам с подозрением на X-сцепленный лимфопролиферативный синдром рекомендуется иммунофенотипирование субпопуляций лимфоцитов для оценки степени выраженности иммунологического дефекта [9, 24].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 3)
Комментарии: У многих пациентов с XЛП1 отмечается снижение (вплоть до нулевых значений) числа B-лимфоцитов, либо значительное снижение переключенных B-клеток памяти (CD19+CD27+IgD-IgM-). Показатели иммунофенотипирования лимфоцитов при XЛП2 не специфичны. Для XЛП3 характерна CD4-лимфопения. Снижение НКТ наиболее характерно для XЛП1, может встречаться при XЛП2, не характерно для XЛП3
- Всем пациентам с подозрением на X-сцепленный лимфопролиферативный синдром рекомендуется определение внутриклеточной экспрессии белков SAP и XIAP методом проточной цитометрии для подтверждения верификации диагноза [4].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 3)
Комментарии: для пациентов с XЛП1 и XЛП2 характерно значительное снижение и полное отсутствие экспрессии белков SAP и XIAP соответственно.
- Пациентам с подозрением на X-сцепленный лимфопролиферативный синдром рекомендуется молекулярно-генетическое исследование соответствующих генов (SH2D1A, XIAP (он же BIRC4), MAGT1) для подтверждения диагноза у пациента и у других членов семьи мужского пола, а также диагностики матерей-носителей [25].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 3)
- Пациентам с подозрением на X-сцепленный лимфопролиферативный синдром рекомендуется проводить ПЦР исследование крови (по показаниям других биологических сред) на определение содержания ДНК вируса Эпштейн-Барр (ЭБВ), цитомегаловируса (ЦМВ), герпеса 6 типа (HHV VI), а также микробиологическое исследование биологических сред при наличии соответствующих очагов инфекции для диагностики сопутствующей инфекции и определения вирусологического статуса пациента [16, 26].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 4)
Комментарии: Серологические исследования у пациентов с ПИДС, в частности с XЛП, мало информативны. Вирусологический статус пациента характеризуется количественным (предпочтительно) или качественным определением вирусов методом полимеразной цепной реакции (ПЦР) в крови, кале, ликворе, бронхо-альвеолярном лаваже, биопсийном материале. В случае XЛП в первую очередь рекомендовано определить количество копий вируса ЭБВ в крови и, по показаниям, в других средах. Учитывая ЦМВ- и HHV VI-ассоциированные ГЛГ рекомендовано также определение количества данных вирусов в крови и других средах. Посевы биоматериала (на флору и грибы) с определением антибиотикочувствительности со слизистых, из очагов инфекции (включая посев крови и мочи при соответствующей симптоматике), а также посевы кала, бронхоальвеолярного лаважа, ликвора и биопсионного материала рекомендовано проводить всегда при наличии инфекционных очагов.
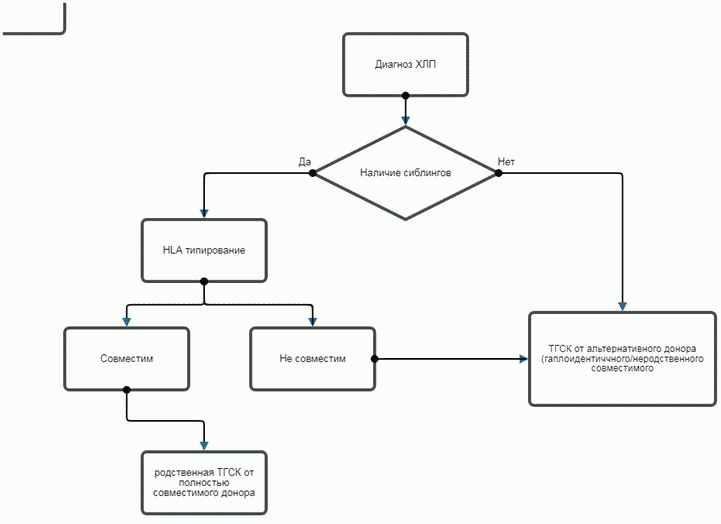
- Пациентам с подозрением на X-сцепленный лимфопролиферативный синдром рекомендуется HLA-типирование для подготовки к пересадке костного мозга, определению совместимости с сиблингами, родителями (при отсутствии сиблингов), или типирование для поиска неродственного донора [6, 27].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 3)
2.4 Инструментальная диагностика
- Всем пациентам с подозрением на X-сцепленный лимфопролиферативный синдром рекомендуется компьютерная томография (КТ) грудной и брюшной полости с контрастным усилением для оценки выраженности лимфопролиферации, исключения лимфомы; магнитно-резонансная терапия (МРТ) головного мозга с контрастным усилением для оценки поражения центральной нервной системы (ЦНС) при ГЛГ, а также для исключения васкулита и его осложнений (тромбоз, аневризма и т.д.) у пациентов с XЛП1 [26, 28].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 3)
- Всем пациентам с подозрением на X-сцепленный лимфопролиферативный синдром рекомендуется костномозговая пункция для подтверждения течения ГЛГ, а также оценки его поражения костного мозга при развитии лимфом [10].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 3)
- Пациентам с подозрением на X-сцепленный лимфопролиферативный синдром, осложненный гемофагоцитозом или васкулитом с поражением ЦНС, рекомендуется люмбальная пункция с цитологическим исследованием ликвора, а также определение содержания белка, глюкозы, для подтверждения или исключения вовлеченности ЦНС [16, 28].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 3)
- Пациентам с подозрением на X-сцепленный лимфопролиферативный синдром рекомендуется по показаниям гастроскопия и колоноскопия с гистологическим исследованием биоптатов желудка и кишечника (при наличии кишечного синдрома) для уточнения характера поражения желудочно-кишечного тракта (ЖКТ) и решения вопроса об иммуносупрессивной терапии и/или хирургическом методе лечения [29].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 3)
3. Лечение
3.1 Консервативное лечение
Цель лечения: стабилизация состояния, контроль развившихся осложнений и предотвращение новых осложнений на период подготовки к ТГСК.
Лечение лимфомы. Проводится по протоколам лечения лимфом, в зависимости от ее типа (см. соответствующие клинические рекомендации).
Лечение гемофагоцитарного лимфогистиоцитоза. Проводится по протоколам лечения ГЛГ, в настоящее время - по протоколу HLH2004 (дексаметазон**, этопозид**, циклоспорин** - см. соответствующие клинические рекомендации). Допускается в качестве дополнительной опции применение антицитокиновой терапии, например, препарат моноклональных антител к человеческому рецептору интерлейкина 6 - #Тоцилизумаб**, а также применение селективного ингибитора JAK-киназы - #Руксолитиниба** - назначение которых должно рассматриваться индивидуально в каждом конкретном случае [30 - 32].
Лечение воспалительного заболевания кишечника. Проводится по протоколам лечения воспалительного заболевания кишечника, в зависимости от гистологической картины (см. соответствующие клинические рекомендации).
- Всем пациентам с XЛП рекомендуется проведение заместительной терапии внутривенными иммуноглобулинами (ВВИГ**) только высокого качества ввиду их вирусной безопасности, в том числе в отношении парвовируса B19, который смертельно опасен для пациентов с ПИД [2].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 4)
Комментарии: терапия проводится пожизненно\до момента восстановления иммунитета после ТГСК в стандартных режимах в дозе 400 - 600 мг/кг веса 1 раз в 3 - 4 недели, с целью поддержания уровня IgG не ниже 6 г\л [2]. В случае, если данная концентрация не достигнута, показано увеличение разовой дозы ВВИГ** до 0,8 - 1 гр/кг или сокращение интервала между введениями препарата до 3-х недель. Применение иммуноглобулина человека нормальный [IgG + IgM + IgA]** для внутривенного введения, а также иммуноглобулина для внутримышечного введения противопоказано из-за его неэффективности у пациентов с данным диагнозом.
- Пациентам с XЛП, осложненным ЭБВ-инфекцией рекомендуется лечение #ритуксимабом** [27].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 3)
Комментарии: Специфические вирусостатики, активные в отношении ЭБВ, на сегодняшний день отсутствуют. Так как ЭБВ инфекция представляет угрозу жизни пациента, при выявлении виремии рекомендовано проведение терапии #ритуксимабом** в дозе 375 мг/м2 1 раз в неделю 4 недели, далее - по необходимости под контролем виремии.
- Пациентам с XЛП с нейтропенией менее < 1,0 x 109/л после исключения гемофагоцитоза и гемобластоза рекомендуется назначение препаратов короткого действия гранулоцитарного колониестимулирующего фактора рекомбинантного (рчГ-КСФ) для достижения и поддержания абсолютного количества нейтрофилов > 1,0 x 109/л [33].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 4)
Комментарии: Дозировка и кратность введения ленограстима и филграстима** подбирается индивидуально.
- Профилактическая противомикробная терапия рекомендуется всем пациентам с XЛП с отягощенным инфекционным анамнезом до момента санации хронических очагов инфекции, не менее 3 месяцев. Выбор препарата рассматривается индивидуально в каждом конкретном случае, в том числе с учетом чувствительности выявленной микрофлоры. Стандартная профилактическая антибактериальная терапия - азитромицин** 10 мг/кг/сут 3 р/нед., также назначается (особенно при наличии нейтропении) профилактическая противогрибковая терапия - (флуконазол** 3 - 8 мг/кг/сут.) [34, 35].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 4)
3.2 Хирургическое лечение
- Пациентам с XЛП рекомендуется по показаниям хирургическое лечение в зависимости от осложнений. При выраженном язвенном поражении кишечника целесообразно рассмотреть вопрос о выведении стомы на время проведения ТГСК и раннего посттрансплантационного периода [29, 36].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 3)
3.3 Трансплантация гематопоэтических стволовых клеток (ТГСК)
- Пациентам с XЛП рекомендуется проведение ТГСК, являющейся единственным куративным методом лечения пациентов с XЛП. Проводится от родственного совместимого, неродственного совместимого или гаплоидентичного донора по методикам, используемым в конкретных центрах [6, 37].
Уровень убедительности рекомендаций B (уровень достоверности доказательств - 2)
4. Реабилитация
- Пациентам с XЛП рекомендуется реабилитация после проведения ТГСК, включающая вакцинацию в соответствии с национальным календарем, при необходимости - консультации с психологом, специалистами лечебной физической культуры [38].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 4)
5. Профилактика
- Всем пациентам с XЛП рекомендуется проводить профилактические мероприятия, включающие медико-генетическое консультирование семей и пренатальную диагностику, которая проводится с помощью молекулярно-генетического исследования биоптата хориона с выявлением мутации соответствующего гена, что позволяет предотвратить рождение других пациентов с данным заболеванием в семьях XЛП [38].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 4)
- Всем пациентам с XЛП рекомендуется диспансерное наблюдение педиатра/терапевта и аллерголога-иммунолога по месту жительства. Профилактическая терапия проводится амбулаторно, длительно - до момента проведения ТГСК. Пациенты и члены их семей должны быть обучены правилам индивидуальной гигиены [38].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 4)
Комментарии:
Лабораторные и инструментальные исследования
- Общий клинический анализ крови (обязателен подсчет лейкоцитарной формулы) проводится 1 раз в 1 мес, по показаниям - чаще.
- Биохимический анализ крови с определением активности печеночных ферментов, C-реактивного белка - 1 раз в 1 мес, по показаниям - чаще.
- Общий анализ мочи - 2 раза в год, по показаниям - чаще
- Электрокардиография (ЭКГ) - 1 раз в год.
- Ультразвуковое исследование (УЗИ) брюшной полости - 1 раз в 3 мес.
- Рентгенография грудной клетки - 1 раз в год.
Периодичность осмотра специалистами, проводившими терапию
Аллерголог-иммунолог осматривает пациента 1 раз в 3 мес.
Периодичность контрольных осмотров специалистами смежных специальностей:
- осмотр гематолога - 1 раз в 3 мес;
- осмотр офтальмолога - 1 раз в 6 мес;
- осмотр стоматолога - 1 раз в год;
- осмотр отоларинголога - 1 раз в год
- осмотр невролога - 1 раз в 6 мес;
Вакцинация до проведения ТГСК не показана.
6. Дополнительная информация, влияющая на течение и исход заболевания
Прогноз
При успешной ТГСК прогноз качества и продолжительности жизни в целом благоприятный, он во многом определяется тяжестью сформировавшихся к моменту трансплантации хронических очагов инфекции и поражения органов. Прогностически неблагоприятным фактором к моменту ТГСК для детей и взрослых пациентов является отсутствие выхода в ремиссию ГЛГ и/или злокачественной лимфопролиферации.
Семейное консультирование и пренатальная диагностика.
Семья пациента XЛП
После постановки пациенту диагноза XЛП рекомендовано срочное обследование всех братьев (по показаниям - двоюродных братьев) пациента, независимо от возраста, так как они могут иметь бессимптомное заболевание, еще не инфицировавшись вирусом ЭБВ. Тем не менее, риск смертельного исхода\тяжелейших осложнений при первом контакте с вирусом настолько велик, что бессимптомным пациентам с XЛП также рекомендовано проведение ТГСК [5, 6].
Пренатальная диагностика показана при всех последующих беременностях матери в данном браке и в других браках (X-сцепленный тип наследования). При X-сцепленном типе наследования рекомендовано тестирование на носительство мутации сестер пациента, всех сестер матери детородного возраста, по показаниям - других родственников женского пола [5, 25].
Пациент XЛП после ТГСК
Риск заболевания у детей пациента составляет менее 0,1%. Все дочери пациента являются носителями мутантного гена, им рекомендовано семейное консультирование [25].
Генная терапия
В настоящее время идут активные клинические исследования, которые дадут возможность рутинного применения генной терапии, в том числе и при XЛП, тем не менее на сегодняшний не является терапевтической модальностью [39].
Дополнительные методы диагностики
Для пациентов с XЛП2 характерно снижение продукции TNF-alpha моноцитами в ответ на стимуляцию NOD2 рецепторов. В связи с этим в качестве дополнительного метода верификации диагноза пациентам с подозрением на X-сцепленный лимфопролиферативный синдром 2 типа рекомендуется определение продукции TNF-alpha моноцитами в ответ на их стимуляцию лигандом NOD2-рецепторов - L18-MDP (в России данное исследование пока не проводится) [12].
7. Организация медицинской помощи
Показания для плановой госпитализации:
1. Динамической контроль состояния при хронических очагах инфекции, с целью проведения инструментальных методов исследования, по показаниям (бронхоальвеолярный лаваж, компьютерная томография и т.д.);
2. Оценка эффективности проводимой иммуносупрессивной терапии (терапия ВЗК, поддержание ремиссии ГЛГ, терапия в рамках протоколов лечения лимфом);
3. Проведение эндоскопического обследования для оценки эффективности терапии ВЗК;
4. Проведение предтрансплантационного обследования.
Показания для экстренной госпитализации:
1. Длительная фебрильная лихорадка;
2. Развитие ИМ;
3. Подозрение на течение васкулита (неврологическая симптоматика, судороги и т.д.);
4. Подозрение на развитие ГЛГ;
5. Подозрение на развитие лимфомы;
6. Обострение или подозрение на манифестацию ВЗК.
Показания к выписке пациента из стационара:
1) Стабилизация состояния;
2) Окончание планового обследования.
Критерии оценки качества медицинской помощи
|
N |
Критерии качества |
Уровень достоверности доказательств |
Уровень убедительности рекомендаций |
|
1. |
Выполнен сбор анамнеза и жалоб, физикальное обследование врачом-гематологом и/или аллергологом-иммунологом |
C |
3 |
|
2. |
Выполнено базовое лабораторное обследование (клинический анализ крови; биохимический анализ крови (мочевина, креатинин, фракции билирубина, аспартатаминотрансфераза, аланинаминотрансфераза, альбумина, лактатдегидрогеназа, щелочная фосфатаза, глюкоза, триглицериды, ферритин, C-реактивный белок, фибриноген) |
C |
3 |
|
3. |
Выполнено инструментальное обследование (компьютерная томография грудной и брюшной полости с контрастным усилением или УЗИ брюшной полости и рентгенография грудной клетки |
C |
3 |
|
4. |
Проведено иммунологическое обследование (определение иммуноглобулинов сыворотки крови IgG, IgA, IgM, иммунофенотипирование лимфоцитов) |
C |
3 |
|
5. |
Проведено вирусологическое исследование для исключения виремии Эпштейн-Барр вируса, цитомегаловируса, вируса герпеса 6 типа с помощью метода ПЦР |
C |
4 |
|
6. |
Проведено молекулярно-генетическое исследование потенциально пораженных генов (SH2D1A, XIAP, MAGT1) |
C |
3 |
|
7. |
При подозрении на гемофагоцитоз выполнена костномозговая пункция |
C |
3 |
|
8. |
Выполнена люмбальная пункция и магнитно-резонансная томография головного мозга (при подозрении на поражение центральной нервной системы) |
C |
3 |
|
9. |
Проведена заместительная терапия препаратами внутривенных иммуноглобулинов |
C |
4 |
|
10. |
Проведена антибактериальная, противогрибковая терапии с профилактической целью на этапе наблюдения при наличии рецидивирующих инфекционных заболеваниях |
C |
4 |
|
11. |
Проведено семейное генетическое консультирование пациента, его непосредственной семьи, а также других родственников с целью информирования их о рисках заболевания у потомства пациента\членов семьи. |
C |
4 |
Список литературы
1. Роппельт А.А., Юхачева Д.В. и др. X-сцепленный лимфопролиферативный синдром 1 и 2 типов//Вопросы гематологии/онкологии и иммунологии в педиатрии. 2016; Т. 15(1). с. 17 - 26.
2. Primary immunodeficiency diseases: A molecular and genetic approach. 3rd edition. Ochs HD, Smith CI, Puck JM, eds. Oxford University press; 2013.
3. Marsh RA, Madden L, Kitchen BJ, Mody R, McClimon B, Jordan MB, et al. XIAP deficiency: a unique primary immunodeficiency best classified as X-linked familial hemophagocytic lymphohistiocytosis and not as X-linked lymphoproliferative disease. Blood. 2010; 116(7): 1079 - 82.
4. Marsh R.A., BleesingJ. J., Filipovich A.H. Using Flow Cytometry to Screen Patients for X-linked Lymphoproliferative Disease Due to SAP Deficiency and XIAP Deficiency. J. Immunol. Methods. 2010; 362(1 - 2): 1 - 9.
5. Иммунология детского возраста. Практическое руководство по детским болезням. Под ред. А.Ю. Щербины и Е.Д. Пашанова. М.: Медпрактика - М; 2006.
6. Booth C., Gilmour K.C., Veys P., Gennery A.R., Slatter M.A., Chapel H., et al. X-linked lymphoproliferative disease due to SAP/SH2D1A deficiency: a multicenter study on the manifestations, management and outcome of the disease. Blood. 2011; 117(1): 53 - 62.
7. Purtilo DT, Grierson HL, Davis JR, Okano M. The X-linked lymphoproliferative disease: from autopsy toward cloning the gene 1975 - 1990. Pediatr Pathol. 1991; 11(5): 685 - 710.
8. Tangye SG. XLP: clinical features and molecular etiology due to mutations in SH2D1A encoding SAP. J Clin Immunol. 2014; 34(7): 772 - 9.
9. Pachlopnik Schmid J, Canioni D, Moshous D, Touzot F, Mahlaoui N, Hauck F, et al. Clinical similarities and differences of patients with X-linked lymphoproliferative syndrome type 1 (XLP-1/SAP deficiency) versus type 2 (XLP-2/XIAP deficiency). Blood. 2011; 117(5): 1522 - 9.
10. Latour S., Aguilar C. XIAP deficiency syndrome in humans. Semin Cell Dev Biol. 2015; 39: 115 - 23.
11. Bertrand M.J., et al. Cellular inhibitors of apoptosis cIAP1 and cIAP2 are required for innate immunity signaling by the pattern recognition receptors NOD1 and NOD2. Immunity. 2009; 30: 789 - 801.
12. Ammann S., et al. A new functional assay for the diagnosis of X-linked inhibitor of apoptosis (XIAP) deficiency. Clinical and Experimental Immunology. 2014; 176: 394 - 400.
13. Yabal M.,  , et al. XIAP restricts TNF- and RIP3-dependent cell death and inflammasome activation. Cell Rep. 2014; 7(6): 1796 - 808.
, et al. XIAP restricts TNF- and RIP3-dependent cell death and inflammasome activation. Cell Rep. 2014; 7(6): 1796 - 808.
14. Ravell J, Chaigne-Delalande B, Lenardo M. X-linked immunodeficiency with magnesium defect, Epstein-Barr virus infection, and neoplasia disease: a combined immune deficiency with magnesium defect. Curr Opin Pediatr. 2014 Dec; 26(6): 713 - 9.
15. Li FY, et al Clinical utility gene card for: X-linked immunodeficiency with magnesium defect, Epstein-Barr virus infection, and neoplasia (XMEN). Eur J Hum Genet. 2015 Jun; 23(6).
16. Aguilar C, Latour S. X-linked inhibitor of apoptosis protein deficiency: more than an X-linked lymphoproliferative syndrome. J Clin Immunol. 2015; 35(4): 331 - 8.
17. Woon S.T. et al. Follicular lymphoma in a X-linked lymphoproliferative syndrome carrier female. Scand J Immunol. 2008; 68(2): 153 - 8.
18. Yang X. et al. A female patient with incomplete hemophagocytic lymphohistiocytosis caused by a heterozygous XIAP mutation associated with non-random X-chromosome inactivation skewed towards the wild-type XIAP allele. J. Clin. Immunol. 2015; 35(3): 244 - 8.
19. Picard C. et al. International Union of Immunological Societies: 2017 Primary Immunodeficiency Diseases Committee Report on Inborn Errors of Immunity s. J. Clin. Immunol. 2017; 38(1): 96 - 128.
20. Worthey EA, Mayer AN, Syverson GD, Helbling D, Bonacci BB, Decker B, et al. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med. 2011; 13(3): 255 - 62.
21. Aguilar C, Lenoir C, Lambert N,  , Brousse N, Canioni D, et al. Characterization of Crohn disease in X-linked inhibitor of apoptosis-deficient male patients and female symptomatic carriers. J Allergy Clin Immunol. 2014; 134(5): 1131 - 41. e9.
, Brousse N, Canioni D, et al. Characterization of Crohn disease in X-linked inhibitor of apoptosis-deficient male patients and female symptomatic carriers. J Allergy Clin Immunol. 2014; 134(5): 1131 - 41. e9.
22. Seemayer TA, Gross TG, Egeler RM, Pirruccello SJ, Davis JR, Kelly CM, et al. X-linked lymphoproliferative disease: twenty-five years after the discovery. Pediatr Res. 1995; 38(4): 471 - 8.
23. Щербина А.Ю. Маски первичных иммунодефицитных состояний: проблемы диагностики и терапии. Российский журнал детской гематологии и онкологии (РЖДГиО). 2016; 3(1): 52 - 58.
24. Rigaud S,  , Lambert N, Pasquier B, Mateo V, Soulas P, et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature. 2006; 444(7115): 110 - 4.
, Lambert N, Pasquier B, Mateo V, Soulas P, et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature. 2006; 444(7115): 110 - 4.
25. Кузьменко Н.Б., Варламова Т.В., Мерсиянова И.В., Райкина Е.В., Бобрынина В.О., Щербина А.Ю. Молекулярно-генетическая диагностика первичных иммунодефицитных состояний. Вопросы гематологии\онкологии и иммунопатологии в педиатрии. 2016; 15(1): 10 - 16.
26. Mischler M, Fleming GM, Shanley TP, Madden L, Levine J, Castle V, et al. Epstein-Barr virus-induced hemophagocytic lymphohistiocytosis and X-linked lymphoproliferative disease: a mimicker of sepsis in the pediatric intensive care unit. Pediatrics. 2007; 119(5): 1212 - 8.
27. Chellapandian D, Das R, Zelley K, Wiener SJ, Zhao H, Teachey DT, et al. Treatment of Epstein Barr virus-induced haemophagocytic lymphohistiocytosis with rituximab-containing chemo-immunotherapeutic regimens. Br J Haematol. 2013; 162(3): 376 - 82.
28. Talaat K. R. et al. Lymphocytic Vasculitis Involving the Central Nervous System Occurs in Patients with X-linked Lymphoproliferative Disease in the Absence of Epstein-Barr Virus Infection. Pediatr Blood Cancer. 2009; 53(6): 1120 - 1123.
29. Speckmanna C., et al. X-linked inhibitor of apoptosis (XIAP) deficiency: The spectrum of presenting manifestations beyond hemophagocytic lymphohistiocytosis. Clinical Immunology. 2013; 149: 133 - 141.
30. Jordan M.B. Emergence of Targeted Therapy for Hemophagocytic Lymphohistiocytosis//The hematologist. 2018; VOL. 15, Issue 2.
31. Horne A.,  , et al. How to Treat Involvement of the Central Nervous System in Hemophagocytic Lymphohistiocytosis? Curr. Treat. Options Neurol. 2017; 19(1): 3.
, et al. How to Treat Involvement of the Central Nervous System in Hemophagocytic Lymphohistiocytosis? Curr. Treat. Options Neurol. 2017; 19(1): 3.
32. Sin J.H., Zangardi M.L. Ruxolitinib for secondary hemophagocytic lymphohistiocytosis: First case report. Hematol. Oncol. Stem Cell Ther. 2017.
33. Деордиева Е.А. Нейтропения в практике детского гематолога/онколога/Е.А. Деордиева, А.Ю. Щербина//Онкогематология. - 2015. - N 1. - С. 46 - 52.
34. Kuruvilla M., de la Morena M.T. Antibiotic prophylaxis in primary immune deficiency disorders. J. Allergy Clin. Immunol. Pract. 2013; 1(6): 573 - 82.
35. Lortholary O., Dupont B. Antifungal prophylaxis during neutropenia and immunodeficiency. Clin. Microbiol. Rev. 1997; 10(3): 477 - 504.
36. Worthey EA, Mayer AN, Syverson GD, Helbling D, Bonacci BB, Decker B, et al. Making a definitive diagnosis: successful clinical application of whole exome sequencing in a child with intractable inflammatory bowel disease. Genet Med. 2011; 13(3): 255 - 62.
37. Balashov D., Shcherbina A., Maschan M., et al. Single-Center Experience of Unrelated and Haploidentical Stem Cell Transplantation with  and CD19 Depletion in Children with Primary Immunodeficiency Syndromes. Biol. Blood Marrow Transplant. 2015; 21(11): 1955 - 62.
and CD19 Depletion in Children with Primary Immunodeficiency Syndromes. Biol. Blood Marrow Transplant. 2015; 21(11): 1955 - 62.
38. Стратегия медико-психолого-социальной реабилитации детей с гематологическими и онкологическими заболеваниями./Н.Н. Володин, В.Н. Касаткин, Г.Я. Цейтлин и др.//Онкогематология. - 2015 - Т. 1 - С. 7 - 15.
39. Booth C., Gaspar H.B., Thrasher A.J. Treating Immunodeficiency through HSC Gene Therapy. Trends Mol Med. 2016; 22(4): 317 - 327.
40.
Приложение А1
СОСТАВ РАБОЧЕЙ ГРУППЫ
Балашов Дмитрий Николаевич - доктор медицинских наук, член Национального общества экспертов в области первичных иммунодефицитов, член Национального общества детских гематологов и онкологов.
Роппельт Анна Артуровна - член Национального общества экспертов в области первичных иммунодефицитов, член Национального общества детских гематологов и онкологов, член Европейского общества иммунодефицитов
Румянцев Александр Григорьевич - доктор медицинских наук, профессор, академик РАМН, президент Национального общества экспертов в области первичных иммунодефицитов, член Национального общества детских гематологов и онкологов, член Европейского общества гематологов.
Щербина Анна Юрьевна - доктор медицинских наук, профессор РАН, исполнительный директор Национального общества экспертов в области первичных иммунодефицитов, член Национального общества детских гематологов и онкологов, член Европейского общества иммунодефицитов.
Конфликт интересов: Щербина А.Ю. осуществляет лекторскую деятельность при поддержке компаний CSL Behring, Kedrion, Biotest, РФарм, являющиеся изготовителями\дистрибьюторами препаратов внутривенных иммуноглобулинов. Не участвовала в принятии окончательной редакции раздела лечения.
Приложение А2
МЕТОДОЛОГИЯ РАЗРАБОТКИ КЛИНИЧЕСКИХ РЕКОМЕНДАЦИЙ
Целевая аудитория данных клинических рекомендаций:
1. Гематологи
2. Аллергологи-иммунологи
3. Педиатры
4. Гастроэнтерология
5. Инфекционисты
6. Терапевты
Таблица П1 - Уровни достоверности доказательств
|
Уровень достоверности |
Источник доказательств |
|
I (1) |
Проспективные рандомизированные контролируемые исследования Достаточное количество исследований с достаточной мощностью, с участием большого количества пациентов и получением большого количества данных Крупные мета-анализы Как минимум одно хорошо организованное рандомизированное контролируемое исследование Репрезентативная выборка пациентов |
|
II (2) |
Проспективные с рандомизацией или без исследования с ограниченным количеством данных Несколько исследований с небольшим количеством пациентов Хорошо организованное проспективное исследование когорты Мета-анализы ограничены, но проведены на хорошем уровне Результаты не презентативны в отношении целевой популяции Хорошо организованные исследования "случай-контроль" |
|
III (3) |
Нерандомизированные контролируемые исследования Исследования с недостаточным контролем Рандомизированные клинические исследования с как минимум 1 значительной или как минимум 3 незначительными методологическими ошибками Ретроспективные или наблюдательные исследования Серия клинических наблюдений Противоречивые данные, не позволяющие сформировать окончательную рекомендацию |
|
IV (4) |
Мнение эксперта/данные из отчета экспертной комиссии, экспериментально подтвержденные и теоретически обоснованные |
Таблица П2 - Уровни убедительности рекомендаций
|
Уровень убедительности |
Описание |
Расшифровка |
|
A |
Рекомендация основана на высоком уровне доказательности (как минимум 1 убедительная публикация I уровня доказательности, показывающая значительное превосходство пользы над риском) |
Метод/терапия первой линии; либо в сочетании со стандартной методикой/терапией |
|
B |
Рекомендация основана на среднем уровне доказательности (как минимум 1 убедительная публикация II уровня доказательности, показывающая значительное превосходство пользы над риском) |
Метод/терапия второй линии; либо при отказе, противопоказании, или неэффективности стандартной методики/терапии. Рекомендуется мониторирование побочных явлений |
|
C |
Рекомендация основана на слабом уровне доказательности (но как минимум 1 убедительная публикация III уровня доказательности, показывающая значительное превосходство пользы над риском) или нет убедительных данных ни о пользе, ни о риске) |
Нет возражений против данного метода/терапии или нет возражений против продолжения данного метода/терапии Рекомендовано при отказе, противопоказании, или неэффективности стандартной методики/терапии, при условии отсутствия побочных эффектов |
|
D |
Отсутствие убедительных публикаций I, II или III уровня доказательности, показывающих значительное превосходство пользы над риском, либо убедительные публикации I, II или III уровня доказательности, показывающие значительное превосходство риска над пользой |
Не рекомендовано |
Порядок обновления клинических рекомендаций
Механизм обновления клинических рекомендаций предусматривает их систематическую актуализацию - не реже чем один раз в три года, а также при появлении новых данных с позиции доказательной медицины по вопросам диагностики, лечения, профилактики и реабилитации конкретных заболеваний, наличии обоснованных дополнений/замечаний к ранее утвержденным КР, но не чаще 1 раза в 6 месяцев.
Приложение А3
СВЯЗАННЫЕ ДОКУМЕНТЫ
Нет
Приложение Б
АЛГОРИТМЫ ВЕДЕНИЯ ПАЦИЕНТА
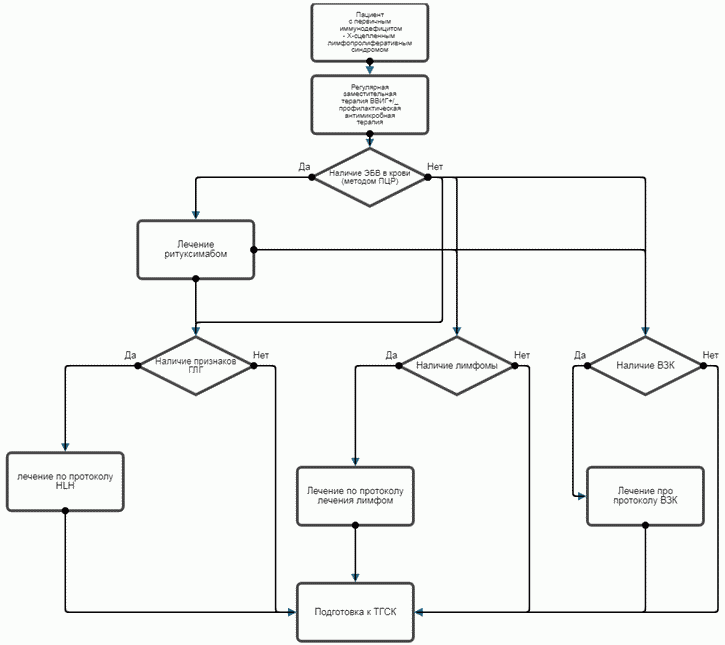
Алгоритм подходов к ТГСК при XЛП
Приложение В
ИНФОРМАЦИЯ ДЛЯ ПАЦИЕНТОВ
X-сцепленный лимфопролиферативный синдром (XЛП) - это комбинированный первичный иммунодефицит (ПИД), характеризующийся атипичной реакцией на инфекцию вирусом Эпштейна-Барр (ЭБВ), вследствие чего развивается гемофагоцитоз, дисгаммаглобулинемией, аутоиммунной патологией, и, в зависимости от типа, злокачественной лимфопролиферацией. На сегодняшний день выделяют 3 типа XЛП с мутациями в генах SH2D1A, XIAP и MAGT1, соответственно.
Высокая предрасположенность к ЭБВ-инфекции, приводящая к развитию фульминантного мононуклеоза, представляющего собой фактически гемофагоцитарный лимфогистиоцитоз, и нарушение количественного состава иммуноглобулинов крови являются общими характеристиками для всех видов XЛП. XЛП 1 типа часто осложняется развитием лимфом, как правило B-клеточных, в то время как для XЛП 2 типа характерно формирование колита, не поддающегося лечению стандартными методами терапии, и нередко требующего хирургического вмешательства для резекции необратимо поврежденного участка кишки.
Клинически стоит обратить внимание на появившийся с раннего возраста жидкий стул, наличие увеличенных лимфоузлов (генерализованная лимфаденопатия или увеличение одной группы лимфоузлов в случае развития лимфомы), эпизоды лихорадки без явного очага инфекции и ответа на антибактериальную терапию. Последнее может сопровождаться увеличением печени/селезенки и являться неполной формой гемофагоцитарного синдрома. Также эпизоды лихорадки и\или субфебрилитета с потами и потерей массы тела могут быть признаками злокачественного заболевания (так называемые "B" симптомы при лимфоме, например). Увеличение печени и селезенки возможно и как проявление хронической ЭБВ инфекции или лимфомы. Появление пятнисто-папулезных высыпаний также должно настораживать в плане ГЛГ.
У пациентов с XЛП 1 и 2 типа могут отмечаться пятнисто-папулезные сыпи, характерные для ГЛГ. При развитии цитопении, в том числе тромбоцитопении, отмечается геморрагическая сыпь.
Лечение пациентов с XЛП представляет собой комплексный подход, включая своевременное и адекватное лечение лимфом, ГЛГ, ЭБВ-инфекции, колита. Однако так как выздоровление от лимфомы или от ГЛГ не гарантирует отсутствие повторных эпизодов, а колит у данных пациент очень плохо поддается иммуносупрессивной терапии, единственным шансом на полное выздоровление в настоящий момент является ТГСК. ТГСК проводится от совместимого брата\сестры, в их отсутствие - от неродственного совместимого донора или от родителей. Исходы ТГСК зависят во многом от имеющегося инфекционного статуса, поражения органов и систем, а также от выхода в ремиссию по ГЛГ и лимфоме к моменту ТГСК.
Риски рождения других мальчиков с XЛП в той же семье составляют 50%. Рекомендовано проведение семейного консультирования и пренатальной\преимплантационной диагностики, для исключения рождения других детей с данным заболеванием.
Приложение Г
Нет