"Клинические рекомендации "Интерферонопатии I типа"

МИНИСТЕРСТВО ЗДРАВООХРАНЕНИЯ РОССИЙСКОЙ ФЕДЕРАЦИИ
КЛИНИЧЕСКИЕ РЕКОМЕНДАЦИИ
ИНТЕРФЕРОНОПАТИИ I ТИПА
МКБ 10: D84.8
Год утверждения (частота пересмотра): 2018 (не реже 1 раза в 3 года)
ID: КР98
URL
Профессиональные ассоциации
- Национальное общество детских онкологов и гематологов - Национальное общество экспертов в области первичных иммунодефицитов - Российская ассоциация аллергологов и клинических иммунологов
Ключевые слова
- Артралгия
- Аутовоспалительный синдром
- васкулит
- Ген ACP5
- Ген ADAR
- Ген DDX58
- Ген IFIH1
- Ген ISG15
- Ген POMP
- Ген PSMB3
- Ген PSMB4
- Ген PSMB8
- Ген PSMB9
- Ген RNASEH2A
- Ген RNASEH2B
- Ген RNASEH2C
- Ген SAMHD1
- Ген SKIV2L
- Ген TMEM173
- Ген TREX1
- Ген USP18
- Гепатомегалия
- Гипергаммаглобулинемия
- Ингибитор JAK-киназы
- Интерлейкин 1
- Интерлейкин 18
- Интерлейкин 6
- Интерферон
- Интерферонопатия
- Лихорадка
- Острофазовые маркеры
- Спленомегалия
- Цитопения
Список сокращений
АВЗ/АВС - аутовоспалительное заболевание/синдром
АлаТ - аланинаминотрансфераза
АНФ - антинуклеарный фактор
АНЦА - Антинейтрофильные Цитоплазматические Антитела
АсаТ - аспартатаминотрансфераза
ВИЧ - вирус иммунодефицита человека
ГКС - глюкокортикостероид
ДМ - дерматомиозит
ДНК - дезоксирибонуклеиновая кислота
ИЛ - интерлейкин
ИФП - интерферонопатии
КМП - пунктат костного мозга (костномозговая пункция)
КТ - компьютерная томография
ЛДГ - лактатдегидрогеназа
ЛП - люмбальная пункция
МКБ-10 - международная классификация болезней 10-го пересмотра
МРТ - магнитно-резонансная томография
ПИДС - первичное иммунодефицитное состояние
РА - ревматоидный артрит
СОЭ - скорость оседания эритроцитов
СРБ - С-реактивный белок
СКВ - системная красная волчанка
УЗИ - ультразвуковое исследование
ФНО - фактор некроза опухоли
ЦИК - циркулирующие иммунные комплексы
ЦНС - центральная нервная система
ЭКГ - электрокардиограмма
ЭхоКГ - эхокардиография ЦНС - центральная нервная система
AGS - Aicardi-Goutieres syndrome, синдром Айкарди-Гутьерес
CANDLE синдром - Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated temperature, хронический атипичный нейтрофильный дерматоз с липодистрофией и подъемами температуры
FDA - Food and Drug Administration, управление по санитарному надзору за качеством пищевых продуктов и медикаментов JASL - Japanese Autoinflammatory Syndrome with Lipodystrophy, Японский аутовоспалительный синдром с липодистрофией
JAK - Janus kinase, янус киназа JMP синдром - Joint contractures, Muscle atrophy, microcytic anemia and Panniculitis-induced lipodystrophy syndrome, Синдром с суставными контрактурами, мышечной атрофией, микроцитарной анемией, индуцированной панникулитом липодистрофией NNS - Nakajo-Nishimura Syndrome, Накайо-Нишимура синдром
NGS - next-generation sequencing, панель генетических мутаций нового поколения
PRAAS - proteasome-associated autoinflammatory syndrome, протеасом-ассоциированный аутовоспалительный синдром
SAVI - STING-Associated Vasculopathy with onset in Infancy, STING-ассоциированная васкулопатия с началом в детском возрасте
SMS - Singleton-Merten syndrome, синдром Синглетон-Мертен
SPENCD - spondyloenchrondro dysplasia, спондилоэнхондродисплазия
IFN - интерферон
Термины и определения
Аутосомно-доминантный тип наследования - тип наследования, при котором одного мутантного аллеля, локализованного в аутосоме, достаточно, чтобы болезнь (или признак) могла быть выражена.
Аутосомно-рецессивный тип наследования - тип наследования признака или болезни, для проявления которых обе копии гена, расположенные на гомологичных аутосомах, должны быть поврежденными.
Дигенный тип наследования - тип наследования, при котором для развития болезни (или признака) необходимо два мутантного аллеля, локализованных в аутосоме.
De novo (мутация de novo) - изменения в гене, произошедшие впервые и только у одного из членов семьи как результат мутации в зародышевой клетке (яйцеклетке или сперматозоиде) у одного из родителей или в уже оплодотворенной яйцеклетке.
Loss of function (LOF, мутации с потерей функции) - мутация, приводящая к снижению или отсутствию функции белка.
Gain of function (GOF, мутации с усилением функции) - мутации, приводящие к аномальному усилению активности белка.
Моногенные заболевания - это гетерогенная группа наследственных заболеваний, различающихся по типу наследования, специфичности мутаций, патогенезу, клинической картине.
Аутовоспаление - воспаление, опосредованное дефектом врожденного звена иммунитета.
Интерферонопатии - это группа заболеваний, обусловленных нарушением индукции, передачи и разрешения интерферон-опосредованного иммунного ответа I типа.
Протеасом-ассоциированные заболевания - это группа заболеваний, относящихся к интерферонопатиям, в основе которых лежит нарушение функции протеасомы.
Убиквитинирование - процесс присоединения одного или нескольких мономеров убиквитина к белку-мишени. Присоединение убиквитина в первую очередь способствует деградации "помеченного" белка в протеасоме, а также влияет на его внутриклеточную локализацию, оказывает воздействие на его активность, способствует или препятствует белок-белковым взаимодействиям.
1. Краткая информация
1.1 Определение
Аутовоспалительные синдромы - это разнородная группа редких, как правило, генетически обусловленных состояний, характеризующихся периодическими эпизодами системного воспаления, проявляющихся лихорадкой и поражением различных органов, нередко имитирующих ревматические и другие заболевания, в отсутствие аутоиммунных или инфекционных причин. По классификации Европейского общества иммунодефицитов аутовоспалительные заболевания (АВЗ) отнесены к первичным иммунодефицитным состояниям, и выделены в отдельную группу.
Интерферонопатии (ИФП) - недавно описанная подгруппа АВЗ, характеризующаяся дисрегуляцией пути интерферонов I типа, и различными аутовоспалительными и аутоиммунными проявлениями. В настоящее время к интерферонопатии относят протеасом-ассоциированные аутовоспалительные синдромы - CANDLE синдром (Chronic Atypical Neutrophilic Dermatosis with Lipodystrophy and Elevated temperature), JMP (Joint contractures, Muscle atrophy, microcytic anemia and Panniculitis-induced lipodystrophy syndrome), NNS (Nakajo-Nishimura Syndrome), JASL (Japanese Autoinflammatory Syndrome with Lipodystrophy), синдром Айкарди-Гутьереса, синдром Синглтон-Мертен, синдром SAVI (STING-Associated Vasculopathy with onset in Infancy) и другие [1, 2, 3].
1.2 Этиология и патогенез
Интерферонопатии являются генетически гетерогенной группой наследственных заболеваний, в основе патогенеза которых лежит дисрегуляция интерферонового пути I типа.
Интерфероны (IFN) - белковые факторы противовирусной защиты, участвующие в регуляции иммунных процессов [4]. В настоящий момент выделяют интерфероны I, II, III типов - в соответствии со способностью взаимодействовать с тремя типами рецепторов. К интерферонам I типа относят 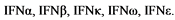 К интерферонам типа II относят
К интерферонам типа II относят ![]() , а к интерферонам III типа - интерфероноподобные цитокины
, а к интерферонам III типа - интерфероноподобные цитокины ![]() (ИЛ-29),
(ИЛ-29), ![]() (ИЛ-28A),
(ИЛ-28A), ![]() (ИЛ-28B). Главная роль в противовирусной защите организма принадлежит интерферонам I типа, в частности
(ИЛ-28B). Главная роль в противовирусной защите организма принадлежит интерферонам I типа, в частности 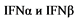 [5].
[5].
Основным продуцентом интерферонов I типа являются плазмоцитоидные дендритные клетки - естественные интерферон-продуцирующие клетки (IPC - interferon-producing cells) [4].
Для синтеза интерферонов в организме необходима активация клеток. В распознании участков чужеродных нуклеиновых кислот вирусов принимают участие Toll-подобные рецепторы (TLR), и RIG-подобные рецепторы (RLR) - RIG-1 и MDA5, а также cGas (циклическая GMP-AMP синтаза) [6]. Основные индукторы синтеза интерферонов I типа - двуспиральная и односпиральная РНК вирусов, а также бактериальная ДНК [4].
RIG-подобные рецепторы распознают двуцепочечные и одноцепочечный вирусные РНК, cGAS (циклическая GMP-AMP синтаза) напротив, распознает двуцепочечные ДНК и РНК: ДНК дуплексы, образующиеся во время репликации ретровирусов, и катализирует синтез cGMP-AMP, который является основным агонистом адапторного белка STING. После связывания с участками РНК RIG-1 и MDA5 связывает белок-адаптер MAVS. Как STING, так и MAVS стимулируют нижестоящие сигнальные каскады, которые включают множественные киназы и, наконец, приводят к фосфорилированию IRF3 и индукции синтеза интерферонов [7, 8], (рис. 1).
Рисунок 1. Схема: РНК-RIG-MAVS и ДНК-GAS-STING путей активации синтеза интерферонов I типа.
Рецептор интерферонов I типа (IFNAR) состоит из двух цепей - IFNAR1 и IFNAR2, с которыми связаны две тирозинкиназы: Tyk2 и Jak1 соответственно. Соединяясь с внутриклеточным доменом рецептора, киназы фосфорилируют друг друга, создавая участок для связывания с транскрипционным фактором STAT2, фосфорилируют его, в результате чего к рецепторному комплексу присоединяется другой фактор - STAT1. Образуется гетеродимер, транслоцирующийся в ядро клеток, где молекулы STAT связываются с интерфероновыми регуляторными факторами с образованием IFN-stimulated gene factor (IFGS3). IFGS3 взаимодействует с ISRE (IFN-stimulated regulatory element), связывание которого необходимо для экспрессии интерферонозависимых генов [4], (рис. 2).
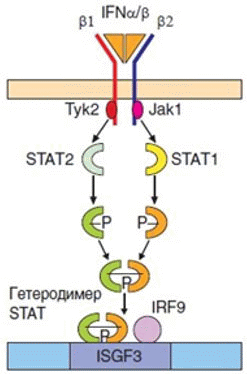
Рисунок 2. Схема передачи сигнала через рецептор интерферонов I типа [4].
Синдром Айкарди-Гутьерес (AGS) - это вид интерферонопатии с частым поражением ЦНС. В настоящий момент известно о 7 генах, мутации в которых приводят к развитию данной патологии, соответственно выделяют 7 типов заболевания. AGS чаще всего наследуется по аутосомно-рецессивному типу; в более редких случаях - аутосомно-доминантно (мутации генов ADAR, TREX1, IFIH1) [10], (таб. 1).
Таблица 1. Классификация синдрома Айкарди-Гутьереса
Тип
Ген
Белок
Год описания первой мутации
Тип наследования
Хромосомная локализация
Тип мутации
I
TREX1
3"-5"экзонуклеаза
2006
АД/АР
3p21.21
Loss of function
II
RNASEH2B
Каталитическая составляющая комплекса РНКазы H2
2006
АР
11q13.1
Loss of function
III
RNASEH2C
Некаталитический компонент комплекса РНКазы H2
2006
АР
19p13.2
Loss of function
IV
RNASEH2A
Некаталитический компонент комплекса РНКазы H2
2006
АР
13q14.3
Loss of function
V
SANHD1
дНТФ трифосфогидролаза-трифосфатаза
2009
АР
20q11.23
Loss of function
VI
ADAR
РНК-специфическая аденозиндезаминаза
2012
АД/АР
1q21.3
Loss of function
VII
IFIH1
MDA5-рецептор
2014
АД
2q24.2
Gain of function
АД - аутосомно-доминантный, АР - аутосомно-рецессивный.
Ген TREX1 расположен на коротком плече 3-й хромосомы и кодирует ядерный белок с 3"-экзонуклеазной активностью. Он прикрепляется к мембране эндоплазматического ретикулума с помощь. C-терминального трансмембранного домена. Этот фермент удаляет ненужные короткие фрагменты молекулы ДНК, которые образуются при репликации генетического материала клетки. Мутации с потерей функции в этом гене приводят к накоплению фрагментов ДНК в цитозоле клетке [10 - 14].
RNASEH2 - единый ферментативный комплекс, состоящий из трех субъединиц, A, B и C, катализирующий расщепление молекул РНК в РНК/ДНК дуплексе посредством гидролитического механизма. В результате мутаций в этих генах с потерей функции происходит накопление ДНК-встроенных рибонуклеотидов, приводящее к повреждению ДНК клетки [15].
Ген SAMHD1 кодирует фермент, представляющий из себя антиретровирусный белок, экспрессируемый в клетках миелоидной линии, который ингибирует раннюю стадию жизненного цикла вируса. Белок превращает дезоксинуклеозидтрифосфаты (dNTP), образующиеся в клетках при репликации ДНК, в неорганический фосфат и 2"-дезоксинуклеозид. Таким образом, в клетке не происходит накопление dNTP до уровня, необходимого для обратной транскрипции и последующей репликации ретровирусов. В результате мутаций с потерей функции в гене происходит накопление фрагментов dNTP [16 - 17].
Ген ADAR кодирует РНК-специфическую аденозиндезаминазу, превращающую аденозин в инозин в двуцепочечных молекулах РНК, что нарушает нормальное связывание цепей по принципу комплементарности, и делает РНК неустойчивой. Инозин структурно подобен гуанину, который приводит к связыванию инозина с цитозином. Двуцепочечные РНК могут образовываться в клетке при взаимодействии смысловых и антисмысловых РНК, является элементом вторичной структуры некоторых РНК (например, тРНК), а также образует геном некоторых РНК-содержащих вирусов. Мутации с потерей функции в этом гене приводят к накоплению в клетке двунитевых РНК [18].
Ген IFIH1 кодирует MDA5-рецептор, относящийся к RIG-подобным рецепторам, который играет важную роль в определении вирусной двухспиральной РНК и активации каскада противовирусных реакций. При связывании лиганда он ассоциируется с митохондриальным противовирусным сигнальным белком (MAVS/IPS1), который запускает два сигнальных пути. Первый - через систему киназ IKK активирует транскрипционный фактор NF-kB, что вызывает активацию клетки и синтез ряда провоспалительных цитокинов. Второй - через адапторный белок TRAF3 и протеинкиназу TBK1 активирует два транскрипционных фактор IRF3 и IRF7, индуцирующих синтез интерферонов бета и альфа соответственно [19].
Синдром Синглтон-Мертена (SMS) - это редкое аутосомно-доминантное заболевание из группы интерферонопатий I типа, в основе которого лежат мутации в генах IFIH1, DDX58, наследуемые по аутосомно-доминантному типу.
Ген DDX58 представляет собой РНК-геликазу, относящийся к RIG-подобным рецепторам, включает MDA5 и LGP2 и играет важную роль в определении вирусной РНК и активации каскада противовирусных реакций. RIG-I и MDA5 участвуют в активации MAVS и запуске противовирусного ответа [20 - 22].
Протеасом-ассоциированные синдромы (PRAAS)
Протеасомы играют важную роль в различных процессах, направленных на поддержание клеточного гомеостаза, поэтому протеасомные заболевания с нарушением функции протеасом отличаются большой вариабельностью течения и последствий, вплоть до очень тяжелых [23].
При PRAAS наблюдается нарушение функции системы "протеасома-иммунопротеасома", что приводит к накоплению убиквитированных белков в различных клетках (B-клетках, фибробластах, CD68-макрофагах, кератиноцитах, клетках волосяных фолликулов и секретирующих клетках потовых желез). Это ведет к усилению фосфорилирования белка p38 и активации системы внутриклеточных сигналов, приводящих к синтезу и секреции провоспалительных цитокинов. В развитие протеасомных заболеваний ключевую роль играет IL-6, гиперпродукция которого провоцирует рецидивирующие воспалительные атаки [4, 23 - 28], (рис. 3 - 4).
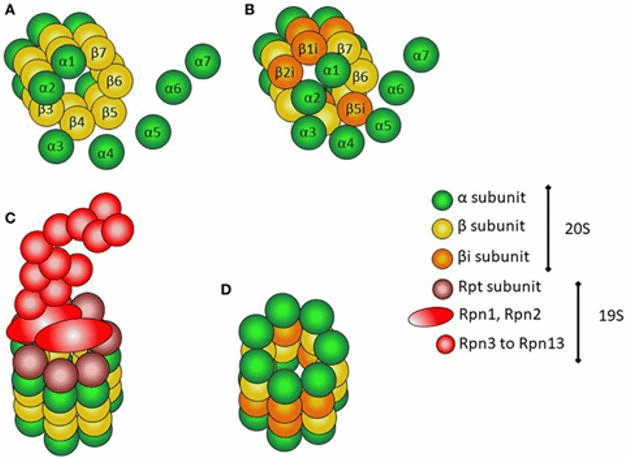
Рисунок 3. Структура протеасомы. (A) Сборка ![]() и
и ![]() субъединиц с образованием 20S ядерного комплекса. (B) Сборка индуцибельных
субъединиц с образованием 20S ядерного комплекса. (B) Сборка индуцибельных 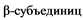 с образованием 20S иммунопротеасомы. (C) 26S протеасома с 20S ядерной частью и регуляторным комплексом 19S. (D) 20S-иммунопротеасома [28].
с образованием 20S иммунопротеасомы. (C) 26S протеасома с 20S ядерной частью и регуляторным комплексом 19S. (D) 20S-иммунопротеасома [28].
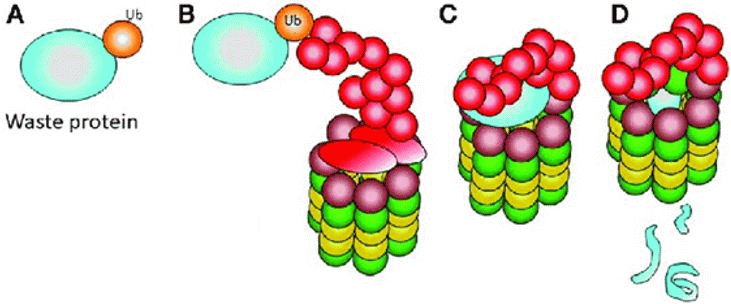
Рисунок 4. Нормальное функционирование протеасомы. (A) Отработанный белок, подлежащий уничтожению, отмечен убиквитином. (B) Комплекс 19S распознает убиквитированный белок. (C) Убиквитинированный белок встраивается в протеосому. (D) Комплекс 20S производит короткие пептиды, легко удаляемые из клетки [28]
При инфицировании клетки вирусом происходит активация центрального белка STING (стимулятор генов интерферона). Когда IFN-рецептор активируется, образованные продукты деградации белков должны быть удалены протеасомой и IFN-индуцированной иммунопротеасомой. Вирусные белки являются субстратом для протеасомной системы. Другие триггеры, вызывающие клеточный стресс, также вызывают высвобождение IFN I типа посредством активации сигнального пути JAK/STAT. Если протеасомная система не работает должным образом, продукты деградации белков накапливаются в клетке и дополнительно помечаются убиквитином (полиубиквитинизация). Накопление полиубиквитинированных белков усугубляет клеточный стресс и способствует продукции IFN I типа, что приводит к порочному кругу и развитию характерной клинической картины [4, 23 - 28] (рис. 5 - 6).
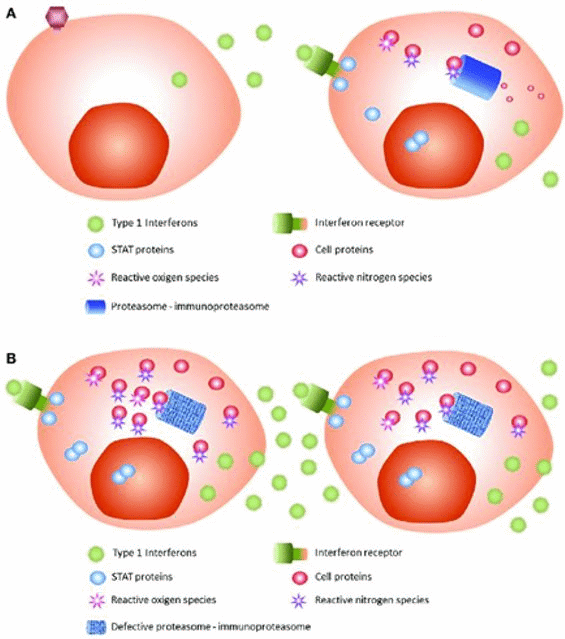
Рисунок 5. (A) Нормальное состояние. (B) При протеасом-ассоциированном заболевании (PRAAS) [28].
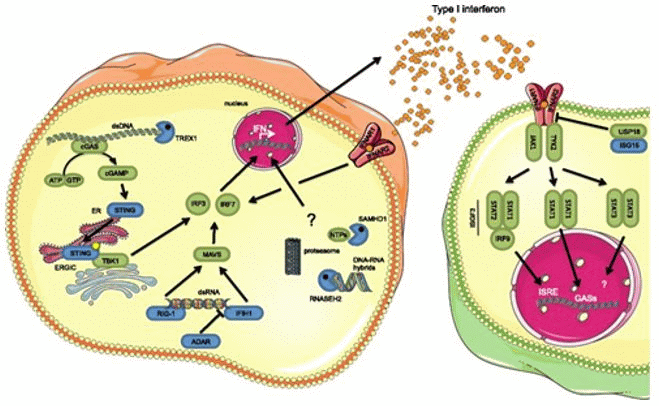
Рисунок 6. Активация интерферонового (IFN) пути [28].
К PRAAS1 относят три нозологии: синдром Накайо-Нишимура (NKJO, японский аутовоспалительный синдром с липодистрофией), хронический атипичный нейтрофильный дерматоз с липодистрофией и подъемами температуры (CANDLE-синдром) и синдром суставных контрактур, мышечной атрофии, микроцитарной анемии и панникулит-ассоциированной липодистрофии (JMP-синдром).
В настоящее время при синдроме PRAAS выявлены мутации в следующих генах: PSMB4, PSMB3, PSMB8, PSMB9. Мутации этих генов наследуются аутосомно-рецессивно. Чаще всего встречаются гомозиготные или компаунд-гетерозиготные мутации в гене PSMB8, а также компаунд-гетерозиготные мутации в гене PSMB4. Интересной особенностью PRAAS является дигенное наследование. Так, описаны случаи заболевания, вызванные комбинацией гетерозиготных мутаций - PSMA3/PSMB8, PSMB9/PSMB4 или PSMB8/PSMB4 [29 - 34].
В июле 2018 года описана мутация нового протеасом-ассоциированного синдрома - PRAAS2. Это редкое заболевание, развивающееся в результате гетерозиготной мутации в гене белка созревания протеасом - POMP, характеризующееся дисрегуляторным расстройством иммунной системы с аутовоспалительным и аутоиммунным компонентами [35 - 36].
POMP - это шаперон, который отвечает за созревание протеасомы 20S и иммунопротеасомы i20s. Он участвует в последовательной сборке в эндоплазматическом ретикулуме  на предварительно сформированных
на предварительно сформированных  кольцах [35 - 36].
кольцах [35 - 36].
В основе патомеханизма PRAAS2 лежит нарушение нонсенс-опосредованного распада мРНК, что позволяет транскрипту мутантного аллеля транслироваться в усеченные белки.
Синдром SAVI (STING-ассоциированная васкулопатия)
SAVI - аутосомно-доминантное заболевание с мутацией в гене TMEM173, кодирующей стимулятор генов интерферона (STING - stimulator of interferon genes), приводящее к аномальному усилению активности белка (GOF - gain of function) [37 - 41].
STING - ключевой адаптерный белок в эндоплазматической сети, который необходим для индукции интерферона бета. Вирусные или двухцепочечные ДНК обнаруживаются в цитозоле с помощью циклической GMP-AMP-синтазы или cGAS-лиганда [42 - 43].
При связывании cGAS циклический гуанозинмонофосфат-аденозинмонофосфат (cGAMP) высвобождается в качестве второго мессенджера, который связывает STING, что приводит к фосфорилированию TANK-связывающей киназы 1 (TBK1) и регуляторного фактора интерферона 3 (IRF3). Затем IRF3 транслоцируется в ядро, что приводит к транскрипции IFNB1 (интерферона ![]() ). Патогенные мутации STING активируют путь, приводящий к транскрипции IFNB1 повышенному уровню его в сыворотке и повышенному фосфорилированию STAT-1 в T и B лимфоцитах [44].
). Патогенные мутации STING активируют путь, приводящий к транскрипции IFNB1 повышенному уровню его в сыворотке и повышенному фосфорилированию STAT-1 в T и B лимфоцитах [44].
Спондилохондродисплазия (SPENCD)
Заболевание наследуется по аутосомно-рецессивному типу, обусловлено мутацией в гене ACP5, кодирующем устойчивую к тартрату щелочную фосфатазу, которая дефосфолирирует и инактивирует остеопонтин - цитокин, регулирующий костеобразование [45, 46]. Остеопонтин способствует прикреплению остеобластов и остеокластов к экстрацеллюлярному матриксу в процессе остеогенеза. Кроме того, остеопонтин необходим для TLR9-зависимой продукции ![]() pDC. Наличие постоянно активированного остеопонтина у пациентов со спондилоэнхондродисплазией обусловливает повышение костной резорбции и сопровождается гиперпродукцией IFNI типа [47].
pDC. Наличие постоянно активированного остеопонтина у пациентов со спондилоэнхондродисплазией обусловливает повышение костной резорбции и сопровождается гиперпродукцией IFNI типа [47].
ISG15 дефицит
Причиной заболевания является аутосомно-рецессивная мутация в гене ISG15, который кодирует убиквитиноподобный протеин. При отсутствии ISG15 в клетках снижается количество убиквитин-специфической протезы 18 (USP18), что приводит к усилению синтеза интерферонов I типа [48, 49].
Дефицит убиквитин-специфической протеазы 18
Заболевание обусловлено аутосомно-рецессивной мутацией в гене USP18, который кодирует убиквитин-специфическую протеазу 18 [50]. Убиквитин-специфическая протеаза 18 (USP18) является ключевым негативным регулятором передачи сигналов IFN I типа, ее дефицит сопровождается гиперпродукцией IFN I типа [51].
Трихогепатоэнтерический синдром 2
Заболевание наследуется по аутосомно-рецессивному типу, обусловлено мутацией в гене SKIV2L, который кодирует белки SKI2 и SKI3, образующие комплекс SKI8. Комплекс SKI8 является кофактором цитозольной экзосомы, учавствующей в деградации аберрантных молекул мРНК [52 - 54].
1.3 Эпидемиология
Интерферонопатии являются редкой патологией, их встречаемость варьирует от 1:10000 до 1:1000000 человек. По данным литературы наиболее часто встречается синдром Айкарди-Гутьереса [55]. Частота недавно описанных заболеваний (например, PRAAS2) не подлежит подсчету, такие случаи в литературе на сегодняшний день единичные [36].
1.4 Кодирование по МКБ 10
Другие иммунодефициты (D84):
D84.8 - Другие уточненные иммунодефицитные нарушения (данный код также используется в КР - Первичный иммунодефицит - X-сцепленный лимфопролиферативный синдром и КР по врожденной нейтропении).
1.5 Классификация
Официально принятой классификации аутовоспалительных синдромов, а также интерферонопатий, в настоящее время, не существует. Однако, исходя из известных механизмов развития заболевания, клинических проявлений и эффективности патогенетической терапии, в международных публикациях моногенные интерферонопатии делятся на следующие подгруппы (табл. 2) [47, 56 - 61]:
Таблица 2. Классификация моногенных форм интерферонопатий
Заболевание
Ген/гены
синдром Айкарди-Гутьерес (AGS)
TREX1, RNASEH2B, RNASEH2C, RNASEH2A, SANHD, ADAR, IFIH1
Синдром Синглтон-Мертена (SMS)
IFIH1, DDX58
Протеосом-ассоциированный периодический синдром (PRAAS)
PSMB4, PSMB3, PSMB8, PSMB, POMP
STING-ассоциированный васкулит с началом в младенчестве (SAVI)
TMEM173
Спондилохондродисплазия (SPENCD)
ACP5
ISG15 дефицит
ISG15
USP18 дефицит (псевдо-TORCH синдром)
USP18
Трихогепатоэнтерический синдром 2
SKIV2L
1.6 Клиническая картина
Клиническая характеристика интерферонопатий очень разнообразна, но можно выделить проявления, характерные для той или иной нозологии, они суммированы в таблице 3 [47, 56, 60 - 62]:
Таблица 3. Клиническая характеристика интерферонопатий
Заболевание
Основные проявления
Синдром Айкарди-Гутьерес (AGS)
Энцефалопатия, мышечная дистония, микроцефалия, кальцификация базальных ганглиев в веществе головного мозга, судороги, лихорадка, повышение острофазовых маркеров крови, цитопения, повышение уровня интерферона в спинно-мозговой жидкости
Синдром Синглтон-Мертена (SMS)
Кардиоваскулярные заболевания с кальцификацией аорты, остеопоретические проявления, зубные и скелетные аномалии, псориатическое поражение кожи
Протеосом-ассоциированный периодический синдром (PRAAS)
Эритематозное поражение кожи, панникулит, липодистрофия, артриты с развитием контрактур суставов, миалгия, гепатомегалия, спленомегалия, кальцификация базальных ганглиев в веществе головного мозга, лихорадка, повышение острофазовых маркеров крови
STING-ассоциированный васкулит с началом в младенчестве (SAVI)
Васкулопатия с формированием дистальной гангрены, некрозов, эритематозная сыпь на лице, кончике носа, ушных раковинах, интерстициальная болезнь легких, артралгия, лихорадка
Спондилохондродисплазия (SPENCD)
Спондилометафизарная дисплазия, низкорослость, кальцификация базальных ганглиев в веществе головного мозга, артропатия, тромбоцитопения, дефицит клеточного и гуморального звена иммунитета
ISG15 дефицит
Кальцификация базальных ганглиев в веществе головного мозга, судороги, микобактериальные инфекции
USP18 дефицит (псевдо-TORCH синдром)
Кровоизлияние и кальцификация в головном мозге, гепатомегалия, тромбоцитопения
Трихогепатоэнтерический синдром 2
Водянистая диарея, ломкие спутанные волосы, поражение печени, умственная отсталость
2. Диагностика
2.1 Жалобы и анамнез
- Рекомендуется всем пациентам с подозрением на интерферонопатию I типа при сборе анамнеза и жалоб у пациента или его родителей для верификации диагноза необходимо выявить [47, 56, 60 - 62]:
- наличие после рождения симптомов, напоминающих течение внутриутробной инфекции, при отсутствии лабораторного подтверждения таковой;
- наличие симптомов угнетения ЦНС в первые сутки после рождения;
- замедление, остановку, регресс нервно-психического развития, отставание в физическом развитии, плохой аппетит и беспокойный сон;
- приобретенную прогрессирующую микроцефалию;
- наличие спастических парезов, мышечной гипотонии, судорожных эпизодов;
- эпизодические подъемы температуры до фебрильных значений либо стойкую субфебрильную лихорадку;
- сыпь с преимущественным поражением кожи пальцев рук, ног, мочек ушей
- диарея;
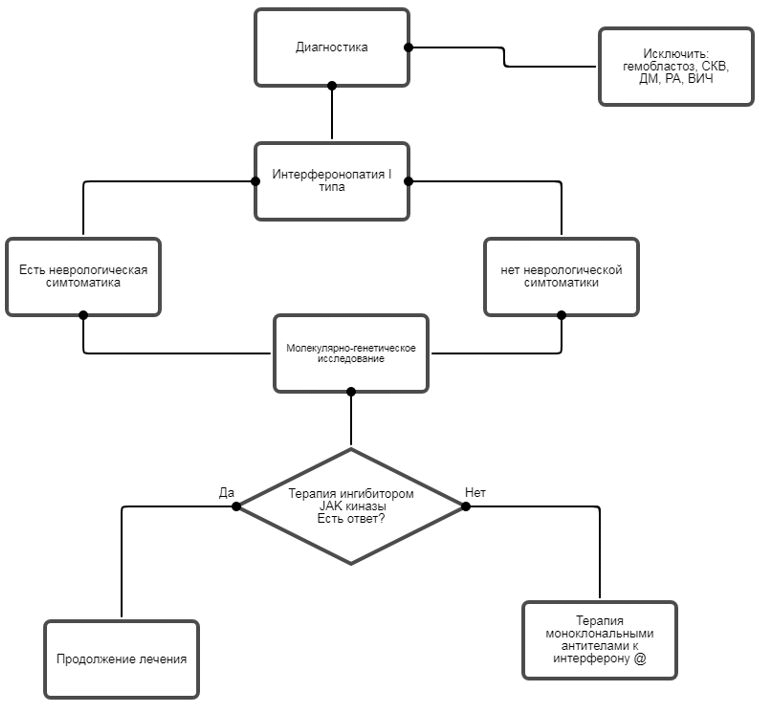
- исключить гемобластоз, СКВ, ДМ, РА, ВИЧ-инфекцию.
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 4)
Комментарии: При сборе семейного анамнеза необходимо обратить внимание на наличие в семье иммунологических, ревматологических, неврологических заболеваний. Обязателен подробный опрос родителей на наличие симптомов заболевания у них, а также у сибсов.
2.2 Физикальное обследование
- Рекомендуется всем пациентам с подозрением на интерферонопатию I типа для верификации диагноза пациента тщательно осматривать кожные покровы, определять мышечный тонус, суставной синдром, исследовать нервную систему с оценкой психомоторного и физического развития. Обязательно проведение термометрии [47, 56, 60 - 62].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 4)
Комментарии:
1. Оценка физического развития - дети могут отставать в физическом и психомоторном развитии. Микроцефалия.
2. Термометрия - возможно повышение температуры тела.
3. Осмотр кожных покровов - важно обратить внимание на наличие кожных высыпаний, сосудистого рисунка, развития подкожно-жировой клетчатки, наличие проявлений панникулита, гангрены, некроза. Липодистрофия верхней половины туловища характерная для протеасом-ассоциированного аутовоспалительного синдрома (PRAAS1, PRAAS3-4). Признаки васкулита, некроза характерны для синдрома SAVI, а также для протеасом-ассоциированного аутовоспалительного синдрома 2 типа. Псориатический дерматит может встречаться при синдроме Айкарди-Гутьерес и синдроме Синглтона-Мертена.
4. Волосы - для пациентов с трихогепатоэнтерическим синдромом характерны ломкие спутанные волосы.
5. Оценка опорно-двигательного аппарата - артралгия, артрит с развитием контрактуры суставов, зубные и скелетные аномалии, миалгия. Дисплазия костной ткани встречается при SPENCD синдроме. Поражение суставов чаще встречается при SMS, PRAAS (кроме PRAAS2).
6. Пальпация групп периферических лимфоузлов - необходимо оценить размеры, консистенцию, болезненность периферических лимфоузлов, возможно развитие локализованной или генерализованной лимфоаденопатии.
7. Пальпация селезенки - выявление спленомегалии. Чаще встречается при AGS, PRAAS.
8. Пальпация печени - выявление гепатомегалии. Чаще встречается при AGS, PRAAS.
9. Оценка центральной нервной системы - диагностика умственной отсталости, мышечной дистонии, энцефалопатии, судорог. Наиболее часто неврологическая симптоматика сопровождает синдром Айкарди-Гутьерес.
2.3 Лабораторная диагностика
- Рекомендуется всем пациентам с подозрением на интерферонопатию I типа выполнить общий анализ крови с целью определения тяжести течения интерферонопатии (наличие лейкоцитоза, тромбоцитопении, ускоренная СОЭ) [60, 61].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 4)
Комментарии: Цитопения (тромбоцитопения, нейтропения, анемия) может встречаться при синдроме Айкарди-Гутьерес, PRAAS2, SPENCD, псевдо-TORCH синдром.
- Рекомендуется всем пациентам с подозрением на интерферонопатию I типа выполнить биохимический анализ крови с обязательным исследованием общего белка, мочевины, креатинина, билирубина, АлаТ, АсаТ, ЛДГ, ЩФ, глюкозы, амилазы, липазы, холестерина с целью определения тяжести течения интерферонопатии и выявления сопутствующей патологии.
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 4)
- Рекомендуется всем пациентам с интерферонопатией I типа исследование сывороточных иммуноглобулинов крови (IgG, IgA, IgM), иммунофенотипирование лимфоцитов, с целью оценки иммунного статуса [60, 61].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 4)
- Рекомендуется всем пациентам с интерферонопатией I типа определение аутоантител (АНФ, антитела к двуспиральной ДНК, Sm антиген, ЦИК, АНЦА) компоненты комплимента (C3, C4), уровень C-реактивного белка для определения уровня аутоантител с целью исключения или подтверждения аутоиммунной агрессии [26, 45, 46, 59, 63].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 3)
- Рекомендуется всем пациентам с подозрением на интерферонопатию определение интерферонового статуса (интерфероны I типа) [47].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 4)
- Рекомендуется всем пациентам с синдромом Айкарди-Гутьереса проведение люмбальной пункции (ЛП) с целью оценки морфологического состава ликвора - при постановке диагноза, далее по необходимости, а также определение уровня ![]() в спинномозговой жидкости [64, 68].
в спинномозговой жидкости [64, 68].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 3)
Комментарии: При цитологическом исследовании возможно определение лимфоцитоза до 200 кл/мм3. Со временем происходит снижение числа лимфоцитов, однако цитоз может сохраняться на протяжении многих лет. На ранних стадиях заболевания может определяться нормальный клеточный состав ликвора при повышенной концентрации ![]() . Нормальное содержание
. Нормальное содержание ![]() < 2 МЕ/мл. Наибольшая активность
< 2 МЕ/мл. Наибольшая активность ![]() в ликворе определяется на ранних стадиях заболевания и обычно выше, чем в периферической крови.
в ликворе определяется на ранних стадиях заболевания и обычно выше, чем в периферической крови.
- Рекомендуется проведение молекулярно-генетического обследования всем пациентам с подозрением на интерферонопатию I типа для верификации диагноза проведение секвенирования нового поколения (NGS) - таргетная панель с включением вышеуказанных генов или полноэкзомное секвенирование [10, 25, 34, 36, 59, 61, 69].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 3)
Комментарии: Фактор наследственности может помочь в установлении диагноза. Например, аутосомно-доминантная мутация в гене TMEM173 является причиной развития STING-ассоциированной васкулопатии (SAVI). Если ребенок болен и у кого-то из членов семьи есть признаки васкулита, поражения легких ассоциированных с мутацией в гене TMEM173, следует предположить наличие мутации в гене TMEM173 и провести молекулярно-генетическое исследование в первые месяцы жизни. Напротив, у пациентов с заболеваниями, которые наследуются по аутосомно-рецессивному типу, например, интерферонопатия вследствие мутаций в генах RNASEH2B, RNASEH2C, RNASEH2A, SAHD1 и других, обычно диагноз обнаруживается среди членов семьи впервые. Клинические данные, указывающие на синдромальную патологию, у данного пациента могут помочь определить, какой именно ген стоит проверить в первую очередь. Например, если ребенка беспокоит лихорадка неинфекционной природы, есть признаки поражения ЦНС, то, следует в план обследования включить гены, отвечающие за развитие синдрома Айкарди-Гутьерес.
2.4 Инструментальная диагностика
- Рекомендуется проведение ультразвукового исследования (УЗИ) брюшной полости, почек, всем пациентам с подозрением на интерферонопатию I типа для верификации диагноза [60, 61].
Комментарии: при УЗ исследовании брюшной полости, почек возможно определение увеличение печени, селезенки, мезентериальных лимфоузлов, патологию почек.
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 4)
- Рекомендуется проведение пациентам компьютерной томографии (КТ) и/или магнитно-резонансную томографию (МРТ) головного мозга для нейровизуализации и определения патологических отклонений [68].
Комментарии: при КТ исследовании возможно определение кальцификатов, чаще всего локализующихся в базальных ганглиях, особенно бледного шара, области таламуса, перивентрикулярных областях. При МРТ исследовании оцениваются изменения белого вещества, атрофия вещества головного мозга, которая может быть прогрессирующей; атрофия мозжечка и атрофия ствола головного мозга, двухсторонний полосатый некроз и внутрицеребральная васкулопатия.
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 4)
- Рекомендуется проведение КТ органов грудной клетки, органов брюшной полости с контрастным усилением всем пациентам с интерферонопатией I типа для верификации диагноза [60, 61].
Комментарии: при КТ исследовании возможно выявление инфекционных очагов, поражение сосудов (например, аневризма), лимфопролиферации, а также с целью исключения неопластического/паранеопластического синдрома.
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 4)
3. Лечение
3.1 Консервативное лечение
Четких алгоритмов лечения пациентов с интерферонопатиями I типа в настоящий момент не разработано ввиду: редкой встречаемости пациентов и относительно недавнего выявления большинства молекулярных причин заболевания; устойчивости к традиционным методам противовоспалительного лечения.
- Рекомендуется пациентам с интерферонопатией I типа терапия ингибитором JAK киназы [79, 81 - 84].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 3)
Комментарии: ингибитор JAKкиназы применяется в дозе 0,3 - 1 мг/кг/сут в 4 - 5 приемов, в таблетированном виде под контролем показателей крови, самочувствия пациента. Стартовая доза для #тофацитиниба** и #руксолитиниба** 0,5 мг/кг/сутки, для #барицитиниба 0,3 мг/кг/сутки. При недостаточной эффективности через 1 неделю применения препарата рекомендовано увеличение дозы под контролем показателей крови (общий анализ крови, биохимия крови), общего самочувствия пациента. Подобранная доза и режим введения используется длительно (пожизненно).
- Рекомендуется пациентам с интерферонопатией I типа терапия моноклональными антителами к интерферону альфа - #сифалимумаб и IFNAR - #анифролумаб при отсутствии эффективности терапии ингибитором JAKкиназы [85, 86].
Уровень убедительности рекомендаций A (уровень достоверности доказательств - 1)
Комментарии: Вопрос о назначении моноклональных антител к интерферону решается индивидуально, с проведением консилиума специалистов, с учетом степени выраженности патологии со стороны других органов и систем. У пациентов с ранним дебютом заболевания, с тяжелыми неврологическими расстройствами, не получающих патогенетическую терапию, плохой прогноз с высоким риском смерти на первом году жизни. Препараты в РФ не зарегистрированы.
- Рекомендуется пациентам с интерферонопатией I типа короткий курс терапии ГКС при ухудшении состояния или с целью купирования основных проявлений [73].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 3)
Комментарии: высокие дозы #ГКС** (1 - 2 мг/кг/сутки по преднизолону**) могут кратковременно улучшить клиническую и лабораторную картину у пациента с интерферонопатией I типа, но впоследствии лечение становится полностью неэффективным.
- Рекомендуется пациентам с интерферонопатиями I типа терапия ингибитором ИЛ6 в качестве комбинации с ингибитором JAKкиназы, монотерапия ингибитора ИЛ6 недостаточна эффективна [44, 70 - 73].
Комментарии: у пациентов с синдромом PRAAS (CANDLE) на терапии ингибитором ИЛ6 (#тоцилизумаб**) в дозе 8 - 12 мг/кг/введение в режиме каждые 2 - 4 недели наблюдается улучшение лабораторных показателей крови с сохранением остальных проявлений заболевания [74, 75].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 4)
- Рекомендуется пациентам с интерферонопатиями I типа терапия #ритуксимабом** в качестве комбинации с ингибитором JAKкиназы, в связи с неэффективностью монотерапии #ритуксимабом** [44, 70 - 73].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 4)
Комментарии: у пациентов с интерферонопатиями I типа с выраженными аутоиммунными проявлениями (положительный титр специфических аутоантител, гипергаммаглобулинемия) в качестве дополнительной терапии рекомендовано применение химерных моноклональных антител, обладающих специфичностью к CD20 антигену в дозе 375 мг/м2/неделю в течение 4 недель [76].
- Не рекомендуется терапия #колхицином (не зарегистрирован в РФ), #азатиоприном**, #метотрексатом**, #циклофосфамидом**, препаратами микофеноловой кислоты, ингибиторами ФНО, ингибиторами ИЛ1 пациентам с интерферонопатиями I типа в связи с их неэффективностью [44, 70 - 73].
Уровень убедительности рекомендаций D (уровень достоверности доказательств - 4)
4. Реабилитация
- Социальная и психологическая реабилитация рекомендуется всем пациентам с интерферонопатиями с целью улучшения качества жизни и возможности социальной адаптации в обществе, а также индивидуальный курс реабилитационных мероприятий, включающий физиотерапию, лечебную физкультуру, массаж [69].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 3)
Комментарии:
- Возможность пребывания в организованном коллективе - после подбора дозы и получения стабильного эффекта от проводимой терапии с развитием лекарственной ремиссии, ограничений для пребывания в детском коллективе нет.
- Возможность путешествий, поездок за границу, пребывания в детском оздоровительном лагере - при стабильном эффекте от проводимой терапии или лекарственной ремиссии, в подобранной дозе ограничений для путешествий нет. В других случаях ограничения могут быть обусловлены неврологическим, физическим, а также клинико-лабораторным статусом пациента.
- Возможность нагрузок и занятий спортом - В состоянии длительной лекарственной ремиссии физические нагрузки определяет врач-реабилитолог, нагрузки не противопоказаны.
- Выбор профессии - в зависимости от степени выраженности заболевания (тяжести проявления), времени начала патогенетической терапии и ответа на проводимую терапию, физических и интеллектуальных способностей зависит выбор будущей профессии.
5. Профилактика
- Рекомендуется всем пациентам с интерферонопатией I типа диспансерное наблюдение педиатра/терапевта по месту жительства с целью оценки состояния здоровья, контроля проводимой терапии, оценки эффективности терапии, после подбора дозы препарата ингибитора JAKкиназы [87 - 89].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 4)
- Рекомендуется диспансерное наблюдение невролога, гематолога, офтальмолога, ортопеда, иммунолога всем пациентам с интерферонопатией после подбора индивидуальной дозы и кратности введения препарата ингибитора JAKкиназы, 1 раз в 1 год с целью оценки состояния здоровья, контроля проводимой терапии, оценки эффективности терапии. При обострении, недостаточной эффективности - чаще [87 - 89].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 4)
- Рекомендуется исследование общего анализа крови с подсчетом лейкоцитарной формулы, определением тромбоцитов, СОЭ всем пациентам с интерферонопатией I типа на фоне патогенетической терапии ингибитором JAKкиназы 1 раз в месяц [60, 61].
Комментарии: При необходимости исследование проводится чаще (на фоне немотивированной лихорадки, ухудшения самочувствия).
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 4)
- Рекомендуется всем пациентам с интерферонопатией I типа проведение планового биохимического анализа крови с обязательным исследованием общего белка, мочевины, креатинина, билирубина, АлаТ, АсаТ, ЛДГ, ЩФ, глюкозы, амилаза, липаза, холестерин, находящимся на терапии JAK киназы 1 раз в 6 месяцев, при необходимости чаще, с целью оценки эффективности терапии [87 - 89].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 3)
- Рекомендуется проведение инструментальных методов исследования (ЭКГ, УЗИ органов брюшной полости, почек, ЭХО-сердца, денситометрия, рентгенография органов грудной клетки, КТ, МРТ) всем пациентам с интерферонопатией I типа с целью оценки общего состояния пациента, наличия или отсутствия хронических очагов инфекции [87 - 89].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 4)
Комментарии:
- ЭКГ - 1 раз в год
- УЗИ брюшной полости, ЭХО-сердца - 1 раз в год
- Рентгенография грудной клетки, КТ, МРТ - по показаниям
- Рекомендуется всем пациентам с интерферонопатией I типа проведение периодических контрольных осмотров специалистами смежных специальностей с целью оценки общего состояния здоровья пациентов, в частности для оценки сопутствующей патологии в составе синдромальной патологии [87 - 89].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 4)
Комментарии:
- Осмотр врача-невролога - 1 раз в год
- Осмотр врача-хирурга - 1 раз в год
- Осмотр врача-стоматолога - 1 раз в год или по показаниям
- Осмотр врача-отоларинголога - 1 раз в год
- Осмотр врача-кардиолога - 1 раз в год
- Осмотр врача-гастроэнтеролога - 1 раз в год
- Осмотр врача-офтальмолога - 1 раз в год
- Осмотр врача-дерматолога - 1 раз в год
- Осмотр врача-ортопеда - 1 раз в год
- Осмотр врача-гематолога - 1 раз в год
При наличии синдромальной патологии возможна более частая консультация специалистов, в зависимости от показаний.
- Рекомендуется всем пациентам с интерферонопатией I типа проводить медико-генетическое консультирование для ознакомления семьи с возможными рисками рождения ребенка с данной патологией, обсуждения вопроса пренатальной/преимплантационной диагностики [44, 59, 61, 69].
Уровень убедительности рекомендаций C (уровень достоверности доказательств - 3)
6. Дополнительная информация, влияющая на течение и исход заболевания
Использования ингибитора JAK киназы во время беременности/в период лактации
Безопасность применения ингибиторов JAKкиназы при беременности не изучена, информации недостаточно, поэтому препарат противопоказан к применению при беременности [90].
Вакцинация
Вакцинация проводится всем пациентам с интерферонопатией (детям и взрослым) при достижении контроля над заболеванием. Интерферонопатии не являются противопоказанием для вакцинации, вакцинацию следует проводить в соответствии с национальным календарем вакцинации, на фоне стабильного состояния пациента, отсутствии катаральных проявлений, отсутствии обострения хронических заболеваний. Рекомендовано заменить ОПВ на ИПВ. Противопоказаний для проведения проба Манту/Диаскин теста нет.
7. Организация медицинской помощи
Показания для плановой госпитализации:
1) Динамический контроль за состоянием при хронических очагах инфекции, с целью проведения инструментальных методов исследования, по показаниям (бронхоальвеолярный лаваж, компьютерная томография, МРТ головного мозга, люмбальная пункция и т.д.);
2) Подбор дозы и частоты введения ингибитора JAKкиназы;
3) недостаточная эффективность или неэффективность проводимой терапии ингибитором JAKкиназы;
Показания для экстренной госпитализации:
1) Фебрильная лихорадка без инфекционного очага на фоне проводимой терапии;
2) прогрессирующая цитопения;
3) прогрессирование неврологической симптоматики, судороги.
Показания к выписке пациента из стационара:
1) Стабилизация состояния;
2) Окончание планового обследования;
3) Подбор дозы и кратности введения ингибитора JAKкиназы.
Критерии оценки качества медицинской помощи
N
Критерии качества
Уровень достоверности доказательств
Уровень убедительности рекомендаций
1
Выполнен осмотр врачом иммунологом и/или педиатром
C
4
2
Выполнен общий анализ крови
C
4
3
Выполнен биохимический анализ крови
C
4
3
Проведено исследование сывороточных иммуноглобулинов крови (IgG, IgA, IgM)
C
4
4
Проведено исследование уровня аутоантител, компонентов комплимента, С-РБ
C
4
5
Проведено определение уровня интерферона a в крови (и ликворе при AGS)
C
4
6
Проведено молекулярно-генетическое исследование: секвенирование нового поколения (NGS) - таргетная панель или полноэкзомное секвенирование
C
4
7
Проведена магнитно-резонансная или компьютерная томография головного мозга
C
4
8
Проведена терапия ингибитором JAKкиназ
C
4
9
Отсутствие осложнений на момент выписки из стационара
C
4
10
На этапе лечения ингибитором JAKкиназ, назначена сопроводительная терапия (неврологического, гастроэнтерологического, дерматологического, гематологического профиля)
C
4
11
На этапе лечения при отсутствии ответа у пациента на ингибиторы JAK, проведена терапия моноклональными антителами к интерферону альфа
A
1
Список литературы
1. Козлова А.Л., Мамзерова Е.С., Новичкова Г.А., Щербина А.Ю. Клинические проявления и терапия криопирин-ассоциированных периодических синдромов. Вопросы гематологии/онкологии и иммунопатологии в педиатрии, 2014; 13: 42 - 48.
2. Fietta P. Autoinflammatory diseases: the hereditary periodic fever syndromes. Acta Biomed. 2004; 75(2): 92 - 99.
3. Gattorno M. Данные конгресса PRES2018 (Pediatric Rheumatology European Society), 5 - 8 September 2018.
4. Ярилин А.А. Иммунология. - М.: ГЭОТАР-Медиа, 2010.
5. Хаитов Р.М. Иммунология: структура и функции иммунной системы: учеб. пособие/Р.М. Хаитов. - М.: ГОЭТАР-Медиа, 2014. - 280 с.
6. Kato H, Oh SW, Fujita T. RIG-I-Like Receptors and Type I Interferonopathies. J Interferon Cytokine Res. 2017 May; 37(5): 207 - 213.
7. Хаитов Р.М. Иммунология: атлас/Р.М. Хаитов, А.А. Ярилин, Б.В. Пинегин. - М.: ГОЭТАР-Медиа, 2011. - 324 с.
8. Zevini A, Olagnier D, Hiscott1 J. Cross-Talk between the Cytoplasmic RIG-I and STING Sensing Pathways // Trends Immunol. 2017 Mar; 38(3): 194 - 205.
9. Miner JJ, Diamond MS. MDA5 and autoimmune disease // Nat Genet. 2014 May; 46(5): 418 - 419.
10. Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1 // Am J Med Genet A. 2015; 2015; 167A: 296 - 312.
11. Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A et al. Mutations in the gene encoding the 3_-5_ DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat. Genet. 2006; 38: 917 - 920.
12. Chowdhury D, Beresford PJ, Zhu P, Zhang D, Sung JS, Demple B, Perrino FW, Lieberman J. The exonuclease TREX1 is in the SET complex and actsin concert with Nm23-H1 to degrade DNA during granzyme A-mediated cell death. Mol. Cell. 2006; 23: 133 - 142.
13. Yang YG, Lindahl T, Barnes DE. TREX1 exonuclease degrades ssDNA to prevent chronic checkpointactivation and autoimmune disease. Cell. 2007; 131: 873 - 886.
14. Stetson DB, Ko JS, Heidmann T, Medzhitov R. TREX1 prevents cell-intrinsic initiation of autoimmunity. Cell. 2008; 134: 587 - 598.
15. Reijns MA, Rabe B, Rigby RE, Mill P, Astell KR, Lettice LA, Boyle S, Leitch A, Keighren M, Kilanowski F, Devenney PS, Sexton D, Grimes G, Holt IJ, Hill RE, Taylor MS, Lawson KA, Dorin JR, Jackson AP. Enzymatic removal of ribonucleotides from DNA is essential for mammalian genome integrity and development. Cell. 2012; 149: 1008 - 1022.
16. Rice GI, Bond J, Asipu A, Brunette RL, Manfield IW et al. Mutations involved in Aicardi-Goutieres syndrome implicate SAM HD1 as regulator of the innate immune response. Nat. Genet. 2009; 41: 829 - 832.
17. Kretschmer S, Wolf C, Konig N, Staroske W, Guck J et al. SAMHD1 prevents autoimmunity by maintaining genome stability. Ann. Rheum. Dis. 2014; 74(3): e17.
18. Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, Walkley CR. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science. 2015; 349(6252): 1115 - 1120.
19. Rice GI, Del Toro DY, Jenkinson EM, Forte GM, Anderson BH, Ariaudo G et al. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat. Genet. 2014; 46: 503 - 509.
20. The American Journal of Human Genetics 96, 266 - 274, February 5, 2015 267 Mutations in DDX58, which Encodes RIG-I, Cause Atypical Singleton-Merten Syndrome Mi-Ae Jang, 1, 16 Eun Kyoung Kim, 2, 16 Hesung Now, 3 Nhung T.H. Nguyen, 3 Woo-Jong Kim, 3 Joo-Yeon Yoo, 3 Jinhyuk Lee, 4, 5 Yun-Mi Jeong, 6.
21. Solis M, Nakhaei P, Jalalirad M, Lacoste J, Douville R, Arguello M, Zhao T, Laughrea M, Wainberg MA, Hiscott J (Feb 2011). "RIG-I-mediated antiviral signaling is inhibited in HIV-1 infection by a protease-mediated sequestration of RIG-I". Journal of Virology. 85 (3): 1224-36.
22. Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ (Aug 2011). "MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response". Cell. 146 (3): 448-61.
23. Федоров Е.С. Протеасомные болезни - новый раздел аутовоспалительной патологии. Современная ревматология. 2013; (4): 38 - 46.
24. Amelia McDermott 1, MA, Jennifer Jacks 2, MD, Marcus Kessler 3, MD, Peter D. Emanuel 4, MD, and Ling Gao 5, MD, PhD Proteasome-associated autoinflammatory syndromes: advances in pathogeneses, clinical presentations, diagnosis, and management.
25. Kim H, Sanchez GA 2, Goldbach-Mansky R Insights from Mendelian Interferonopathies: Comparison of CANDLE, SAVI with AGS, Monogenic Lupus. J Mol Med (Berl). 2016 Oct; 94(10): 1111 - 1127. Epub 2016 Sep 27.
26. Kretschmer S, Lee-Kirsch MA. Type I interferon-mediated autoinflammation and autoimmunity. Curr Opin Immunol. 2017 Dec; 49: 96 - 102. doi: 10.1016/j.coi.2017.09.003.
27. Stefano Volpi, Paolo Picco, Roberta Caorsi, Fabio Candotti and Marco Gattorno. Type I interferonopathies in pediatric rheumatology. Pediatr Rheumatol Online J. 2016 Jun 4; 14(1): 35. doi: 10.1186/s12969-016-0094-4.
28. Torrelo A. CANDLE Syndrome As a Paradigm of Proteasome-Related Autoinflammation. Front. Immunol. 8:927. doi: 10.3389/fimmu.2017.00927
29. Torrelo A, Patel S, Colmenero I, Gurbindo D, Lendinez F, Hernandez A, Lopez-Robledillo JC, Dadban A, Requena L, Paller AS. Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome. J. Am. Acad. Dermatol. 2010; 62: 489 - 495.
30. Arima K, Kinoshita A, Mishima H, Kanazawa N, Kaneko T,Mizushima T et al. Proteasome assembly defect due to a proteasome subunit beta type 8 (PSMB8) mutation causes the autoinflammatory disorder, Nakajo-Nishimura syndrome. Proc. Natl. Acad. Sci. USA. 2011; 108: 14914 - 14919.
31. Kitamura A, Maekawa Y, Uehara H, Izumi K, Kawachi I, Nishizawa M, Toyoshima Y et al. A mutation in the immunoproteasome subunit PSMB8 causes autoinflammation and lipodystrophy inhumans. J. Clin. Invest. 2011; 121: 4150 - 4160.
32. Liu Y, Ramot Y, Torrelo A, Paller AS, Si N, Babay S, Kim PW, Sheikh A, Lee CC et al. Mutations in proteasome subunit beta type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Rheumatism. 2012; 64: 895 - 907.
33. Cavalcante MP, Brunelli JB, Miranda CC, Novak GV, Malle L, Aikawa NE, Jesus AA, Silva CA. CANDLE syndrome: chronic atypical neutrophilic dermatosis with lipodystrophy andelevated temperature-a rare case with a novel mutation. Eur. J. Pediatr. 2016; 175: 735 - 740.
34. Brehm A, Liu Y, Sheikh A, Marrero B, Omoyinmi E, Zhou Q, Montealegre G, Biancotto A et al. Additive loss-of-function proteasome subunit mutationsin CANDLE/PRAAS patients promote type I IFN production. J. Clin. Invest. 2015; 125: 4196 - 4211.
35. Brehm, A., Liu, Y., Sheikh, A., Marrero, B., Omoyinmi, E., Zhou, Q., Montealegre, G., Biancotto, A., Reinhardt, A., Almeida de Jesus, A., Pelletier, M., Tsai, W. L., and 31 others. Additive loss-of-function proteasome subunit mutations in CANDLE/PRAAS patients promote type I IFN production. J. Clin. Invest. 125: 4196 - 4211, 2015. Note: Erratum: J. Clin. Invest. 126: 795 only, 2016.
36. Poli, M. C., Ebstein, F., Nicholas, S. K., de Guzman, M. M., Forbes, L. R., Chinn, I. K., Mace, E. M., Vogel, T. P., Carisey, A. F., Benavides, F., Coban-Akdemir, Z. H., Gibbs, R. A., and 16 others. Heterozygous truncating variants in POMP escape nonsense-mediated decay and cause a unique immune dysregulatory syndrome. Am. J. Hum. Genet. 102: 1126 - 1142, 2018.
37. Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Montealegre Sanchez GA, Tenbrock K, Wittkowski H, Jones OY, Kuehn HS, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. 2014; 371: 507 - 518.
38. Jeremiah N, Neven B, Gentili M, Callebaut I, Maschalidi S, Stolzenberg MC, Goudin N, Fremond ML, Nitschke P, Molina TJ, et al. Inherited STING-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations. J Clin Invest. 2014; 124: 5516 - 5520.
39. Omoyinmi E, Melo Gomes S, Nanthapisal S, Woo P, Standing A, Eleftheriou D, Klein N, Brogan PA. Stimulator of interferon genes-associated vasculitis of infancy. Arthritis Rheumatol. 2015; 67: 808.
40. Munoz J, Rodiere M, Jeremiah N, Rieux-Laucat F, Oojageer A, Rice GI, Rozenberg F, Crow YJ, Bessis D. Stimulator of interferon genes-associated vasculopathy with onset in infancy: a mimic of childhood Granulomatosis with Polyangiitis. JAMA Dermatol. 2015; 151: 872 - 877.
41. Chia J, Eroglu FK, Ozen S, Orhan D, Montealegre-Sanchez G, de Jesus AA, Goldbach-Mansky R, Cowen EW. Failure to thrive, interstitial lung disease, and progressive digital necrosis with onset in infancy. J Am Acad Dermatol. 2016; 74: 186 - 189.
42. Burdette DL, Vance RE. STING and the innate immune response to nucleic acids in the cytosol. Nat Immunol. 2013; 14: 19 - 26.
43. Keating SE, Baran M, Bowie AG. Cytosolic DNA sensors regulating type I interferon induction. Trends Immunol. 2011; 32: 574 - 581.
44. Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Montealegre Sanchez GA, Tenbrock K, Wittkowski H, Jones OY, Kuehn HS, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. 2014; 371: 507 - 518.
45. Briggs TA, Rice GI, Daly S, Urquhart J et al. Tartrate-resistant acid phosphatase deficiency causes a bone dysplasia with autoimmunity and a type I interferon expression signature. Nat. Genet. 2011; 43: 127 - 131.
46. Lausch E, Janecke A, Bros M, Trojandt S, Alanay Y et al. Genetic deficiency of tartrate-resistant acid phosphatase associated with skeletal dysplasia, cerebral calcifications and autoimmunity. Nat. Genet. 2011; 43: 132 - 137.
47. Lee-Kirsch MA. The type I interferonopathies. Ann. Rev. Med. 2017; 68.
48. Bogunovic D, Byun M, Durfee LA, Abhyankar A et al. Mycobacterial disease and impaired IFN-gamma immunity in humans with inherited ISG15 deficiency. Science. 2012; 337: 1684 - 1688.
49. Zhang X, Bogunovic D, Payelle-Brogard B, Francois-Newton V et al. Human intracellular ISG15 prevents interferon-a/b over-amplification and auto-inflammation. Nature. 2015; 517 (7532): 89 - 93.
50. Meuwissen MEC, Schot R, Buta S, Oudesluijs G, Tinschert S et al. Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome. J. Exp. Med. 2016; 213: 1163 - 1174.
51. Anja Basters, Klaus-Peter Knobeloch,  USP18 - a multifunctional component in the interferon response. Bioscience Reports Nov 16, 2018.
USP18 - a multifunctional component in the interferon response. Bioscience Reports Nov 16, 2018.
52. Fabre A, Charroux B, Martinez-Vinson C, et al. SKIV2L mutations cause syndromic diarrhea, or trichohepatoenteric syndrome. Am J Hum Genet 2012; 90: 689-92.
53. Fabre A, Martinez-Vinson C, Goulet O, et al. Syndromic diarrhea/Tricho-hepato-enteric syndrome. Orphanet J Rare Dis 2013; 8: 5.
54. Eckard SC, Rice GI, Fabre A, et al. The SKIV2L RNA exosome limits activation of the RIG-I-like receptors. Nat Immunol 2014; 15: 839-45.
55. https://www.orpha.net.
56. Bienias M,  Griep C, Wolf C, Kretschmer S, Kind B,
Griep C, Wolf C, Kretschmer S, Kind B,  Berner R, Lee-Kirsch MA. Therapeutic Approaches to Type I Interferonopathies. Curr Rheumatol Rep. 2018 Apr 20; 20(6): 32.
Berner R, Lee-Kirsch MA. Therapeutic Approaches to Type I Interferonopathies. Curr Rheumatol Rep. 2018 Apr 20; 20(6): 32.
57. Davidson S, Steiner A, Harapas CR, Masters SL. An Update on Autoinflammatory Diseases: Interferonopathies. Curr Rheumatol Rep. 2018 May 30; 20(7): 38.
58. Kato H, Oh SW, Fujita T. RIG-I-Like Receptors and Type I Interferonopathies. J Interferon Cytokine Res. 2017 May; 37(5): 207 - 213.
59. Kretschmer S, Lee-Kirsch MA. Type I interferon-mediated autoinflammation and autoimmunity. Curr Opin Immunol. 2017 Dec; 49: 96 - 102.
60. Eleftheriou D, Brogan PA. Genetic interferonopathies: An overview. Best Pract Res Clin Rheumatol. 2017 Aug; 31(4): 441 - 459.
61. Kim H, Sanchez GA, Goldbach-Mansky R. Insights from Mendelian Interferonopathies: Comparison of CANDLE, SAVI with AGS, Monogenic Lupus. J Mol Med (Berl). 2016 Oct; 94(10): 1111 - 1127.
62. Torrelo A, Patel S, Colmenero I, Gurbindo D,  et al. Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome. J Am Acad Dermatol (2010) 62: 489-95.
et al. Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature (CANDLE) syndrome. J Am Acad Dermatol (2010) 62: 489-95.
63. Khamashta M, Merrill JT, Werth VP, Furie P, et al. Sifalimumab, an  monoclonal antibody, in moderate to severe systemic lupus erythematosus: a randomised, double-blind, placebo-controlled study // Ann Rheum Dis. 2016 Nov; 75(11): 1909 - 1916.
monoclonal antibody, in moderate to severe systemic lupus erythematosus: a randomised, double-blind, placebo-controlled study // Ann Rheum Dis. 2016 Nov; 75(11): 1909 - 1916.
64. Rice G, Forte GMA, Szynkiewicz M, Chase DS et al. Assessment of interferon-related biomarkers in  syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1 and ADAR: a case-control study // Lancet Neurol. 2013 Dec; 12(12): 1159 - 1169.
syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1 and ADAR: a case-control study // Lancet Neurol. 2013 Dec; 12(12): 1159 - 1169.
65. Bennett L, Palucka AK, Arce E, et al. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med. 2003; 197(6): 711-23.
66. Baechler EC, Batliwalla FM, Karypis G, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci USA. 2003; 100(5): 2610-5.
67. Baechler EC, Bauer JW, Slattery CA, et al. An interferon signature in the peripheral blood of dermatomyositis patients is associated with disease activity. Mol Med. 2007; 13(1 - 2): 59 - 68.
68. Rice G, Patrick T, Parmar R, Taylor CF et al. Clinical and molecular phenotype of Aicardi-Goutieres syndrome // Am J Hum Genet. 2007b; 81: 713-25.
69. Брюханова Н.О., Жилина С.С., Айвазян С.О., Ананьева Т.В., Беленикин М.С., Кожанова Т.В., Мещерякова Т.И., Зинченко Р.А., Мутовин Г.Р., Заваденко Н.Н. Синдром Айкарди-Гутьерес у детей с идиопатической эпилепсией. Российский вестник перинатологии и педиатрии. 2016; 61(2): 68 - 75.
70. Jeremiah N, Neven B, Gentili M, Callebaut I, Maschalidi S, Stolzenberg MC, Goudin N, Fremond ML, Nitschke P, Molina TJ, et al. Inherited STING-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations. J Clin Invest. 2014; 124: 5516 - 5520.
71. Omoyinmi E, Melo Gomes S, Nanthapisal S, Woo P, Standing A, Eleftheriou D, Klein N, Brogan PA. Stimulator of interferon genes-associated vasculitis of infancy. Arthritis Rheumatol. 2015; 67: 808.
72. Munoz J, Rodiere M, Jeremiah N, Rieux-Laucat F, Oojageer A, Rice GI, Rozenberg F, Crow YJ, Bessis D. Stimulator of interferon genes-associated vasculopathy with onset in infancy: a mimic of childhood Granulomatosis with Polyangiitis. JAMA Dermatol. 2015; 151: 872 - 877.
73. Chia J, Eroglu FK, Ozen S, Orhan D, Montealegre-Sanchez G, de Jesus AA, Goldbach-Mansky R, Cowen EW. Failure to thrive, interstitial lung disease, and progressive digital necrosis with onset in infancy. J Am Acad Dermatol. 2016; 74: 186 - 189.
74. J Kluk, M Rustin, PA Brogan, E Omoyinmi, DM Rowczenio, LC Willcocks, L Melly, and HJ Lachmann. Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature syndrome: a report of a novel mutation and review of the literature. Br J Dermatol. 2014 Jan; 170(1): 215 - 217.
75. Cavalcante MP, Brunelli JB, Miranda CC, Novak GV, Malle L, Aikawa NE, Jesus AA, Silva CA. CANDLE syndrome: chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature-a rare case with a novel mutation. Eur J Pediatr. 2016 May; 175(5): 735-40.
76. Midaglia L, Mora L, Mulero P, Sastre-Garriga J, Montalban X. Rituximab: its efficacy, effectiveness and safety in the treatment of multiple sclerosis. Rev Neurol. 2018 Jan 1; 66(1): 25 - 32.
77.  Fiehn C, Wolf C, Schuster M, Cura Costa E,
Fiehn C, Wolf C, Schuster M, Cura Costa E,  et al. Familial chilblain lupus due to a gain-of-function mutation in STING // Ann Rheum Dis. 2017; 76(2): 468 - 472.
et al. Familial chilblain lupus due to a gain-of-function mutation in STING // Ann Rheum Dis. 2017; 76(2): 468 - 472.
78. Rodero MP,  Rice GI, Neven B, Crow YJ. JAK inhibition in STING-associated interferonopathy. // Ann Rheum Dis 2016; 75(12): e75-5.
Rice GI, Neven B, Crow YJ. JAK inhibition in STING-associated interferonopathy. // Ann Rheum Dis 2016; 75(12): e75-5.
79. Kubo S, Yamaoka K, Kondo M, Yamagata K, Zhao J, Iwata S, Tanaka Y. The JAK inhibitor, tofacitinib, reduces the T cell stimulatory capacity of human monocyte-derived dendritic cells. Ann Rheum Dis. 2014 Dec; 73(12).
80. Li Y, Wilson HL, Kiss-Toth E. Regulating STING in health and disease. J Inflamm (Lond). 2017 Jun 7; 14: 11.
81. Kothur K, Bandodkar S, Chu S, Wienholt L, Johnson A, Barclay P, Brogan PA, Rice GI, Crow YJ, Dale RC. An open-label trial of JAK 1/2 blockade in progressive IFIH1-associated neuroinflammation // Neurology. 2018 Feb 6; 90(6): 289 - 291.
82. Sanchez GAM, Reinhardt A, Ramsey S, Wittkowski H et al. JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies // J Clin Invest. 2018 Jul 2; 128(7): 3041 - 3052.
83. Hoffman HM, Broderick L. JAK inhibitors in autoinflammation. J Clin Invest. 2018 Jul 2; 128(7): 2760 - 2762. doi: 10.1172/JCI121526. Epub 2018 Jun 11.
84. Kim H, Brooks KM, Tang CC, Wakim P, Blake M, Brooks SR, Montealegre Sanchez GA, de Jesus AA, Huang Y, Tsai WL, Gadina M, Prakash A, Janes JM, Zhang X, Macias WL, Kumar P, Goldbach-Mansky R. Pharmacokinetics, Pharmacodynamics, and Proposed Dosing of the Oral JAK1 and JAK2 Inhibitor Baricitinib in Pediatric and Young Adult CANDLE and SAVI Patients. Clin Pharmacol Ther. 2018 Aug; 104(2): 364 - 373. doi: 10.1002/cpt.936. Epub 2017 Dec 8.
85. Furie R, Khamashta M, Merrill JT, Werth VP et al. Investigators Anifrolumab, an Anti-Interferon-a Receptor Monoclonal Antibody, in Moderate-to-Severe Systemic Lupus Erythematosus // Arthritis Rheumatol. 2017 Feb; 69(2): 376 - 386.
86. Khamashta M, Merrill JT, Werth VP, Furie P, et al. Sifalimumab, an  monoclonal antibody, in moderate to severe systemic lupus erythematosus: a randomised, double-blind, placebo-controlled study // Ann Rheum Dis. 2016 Nov; 75(11): 1909 - 1916.
monoclonal antibody, in moderate to severe systemic lupus erythematosus: a randomised, double-blind, placebo-controlled study // Ann Rheum Dis. 2016 Nov; 75(11): 1909 - 1916.
87. Практическое руководство по детским болезням. Под ред. В.Ф. Кококлиной и А.Г. Румянцева. Иммунология детского возраста. Под ред. А.Ю. Щербины и Е.Д. Пашанова. Медпрактика-М, 2006.
88. Европейский регистр аутовоспалительных заболеваний. Электронный ресурс. Доступ: https://www.printo.it/eurofever/.
89. Н.Т. Ватутин, А.С. Смирнова, М.А. Эль-Хатиб. Семейная средиземноморская лихорадка: обзор рекомендаций EULAR, 2016. Архивъ внутренней медицины, N 6, 2016: 5 - 11.
90. Gerosa M, Argolini LM, Artusi C, Chighizola CB. The use of biologics and small molecules in pregnant patients with rheumatic diseases. Expert Rev Clin Pharmacol. 2018 Oct; 11(10): 987 - 998.
Приложение А1
СОСТАВ РАБОЧЕЙ ГРУППЫ
1. Козлова Анна Леонидовна - кандидат медицинских наук, член Национального общества экспертов в области первичных иммунодефицитов, член Национального общества детских гематологов и онкологов.
2. Румянцев Александр Григорьевич - доктор медицинских наук, профессор, академик РАМН, президент Национального общества экспертов в области первичных иммунодефицитов, член Национального общества детских гематологов и онкологов, член Европейского общества гематологов.
3. Щербина Анна Юрьевна - доктор медицинских наук, профессор РАН, исполнительный директор Национального общества экспертов в области первичных иммунодефицитов, член Национального общества детских гематологов и онкологов, член Европейского общества иммунодефицитов.
4. Латышева Татьяна Валерьевна - доктор медицинских наук, профессор, член президиума Российской ассоциации аллергологов и клинических иммунологов.
Все члены Рабочей группы подтвердили отсутствие финансовой поддержки/конфликта интересов.
Приложение А2
МЕТОДОЛОГИЯ РАЗРАБОТКИ КЛИНИЧЕСКИХ РЕКОМЕНДАЦИЙ
Целевая аудитория данных клинических рекомендаций:
1. врач-гематолог
2. врач аллерголог-иммунолог
3. врач-педиатр
4. врач-терапевт
5. Врач общей практики (семейный врач)
Таблица П1 - Уровни достоверности доказательств
Уровень достоверности
Источник доказательств
I (1)
Проспективные рандомизированные контролируемые исследования
Достаточное количество исследований с достаточной мощностью, с участием большого количества пациентов и получением большого количества данных
Крупные мета-анализы
Как минимум одно хорошо организованное рандомизированное контролируемое исследование
Репрезентативная выборка пациентов
II (2)
Проспективные с рандомизацией или без исследования с ограниченным количеством данных
Несколько исследований с небольшим количеством пациентов
Хорошо организованное проспективное исследование когорты
Мета-анализы ограничены, но проведены на хорошем уровне
Результаты не презентативны в отношении целевой популяции
Хорошо организованные исследования "случай-контроль"
III (3)
Нерандомизированные контролируемые исследования
Исследования с недостаточным контролем
Рандомизированные клинические исследования с как минимум 1 значительной или как минимум 3 незначительными методологическими ошибками
Ретроспективные или наблюдательные исследования
Серия клинических наблюдений
Противоречивые данные, не позволяющие сформировать окончательную рекомендацию
IV (4)
Мнение эксперта/данные из отчета экспертной комиссии, экспериментально подтвержденные и теоретически обоснованные
Таблица П2 - Уровни убедительности рекомендаций
Уровень убедительности
Описание
Расшифровка
A
Рекомендация основана на высоком уровне доказательности (как минимум 1 убедительная публикация I уровня доказательности, показывающая значительное превосходство пользы над риском)
Метод/терапия первой линии; либо в сочетании со стандартной методикой/терапией
B
Рекомендация основана на среднем уровне доказательности (как минимум 1 убедительная публикация II уровня доказательности, показывающая значительное превосходство пользы над риском)
Метод/терапия второй линии; либо при отказе, противопоказании, или неэффективности стандартной методики/терапии. Рекомендуется мониторирование побочных явлений
C
Рекомендация основана на слабом уровне доказательности (но как минимум 1 убедительная публикация III уровня доказательности, показывающая значительное превосходство пользы над риском или
нет убедительных данных ни о пользе, ни о риске)
Нет возражений против данного метода/терапии или нет возражений против продолжения данного метода/терапии
Рекомендовано при отказе, противопоказании, или неэффективности стандартной методики/терапии, при условии отсутствия побочных эффектов
D
Отсутствие убедительных публикаций I, II или III уровня доказательности, показывающих значительное превосходство пользы над риском, либо убедительные публикации I, II или III уровня доказательности, показывающие значительное превосходство риска над пользой
Не рекомендовано
Порядок обновления клинических рекомендаций
Механизм обновления клинических рекомендаций предусматривает их систематическую актуализацию - не реже чем один раз в три года или при появлении новой информации о тактике ведения пациентов с данным заболеванием. Решение об обновлении принимает МЗ РФ на основе предложений, представленных медицинскими некоммерческими профессиональными организациями. Сформированные предложения должны учитывать результаты комплексной оценки лекарственных препаратов, медицинских изделий, а также результаты клинической апробации.
Приложение А3
СВЯЗАННЫЕ ДОКУМЕНТЫ
Нет.
Приложение Б
АЛГОРИТМЫ ВЕДЕНИЯ ПАЦИЕНТА
Ведение пациентов с подозрением на интерферонопатию I типа
Приложение В
ИНФОРМАЦИЯ ДЛЯ ПАЦИЕНТОВ
Термин интерферонопатия описывает ситуацию, когда в организме пациента происходит избыточная продукция интерферонов I типа. В норме интерфероны защищают организм от вирусных инфекций, но избыточное количество интерферонов может вызывать различные воспалительные проявления.
Интерферонопатии - это клинически разнообразная группа редких заболеваний, в основе которых лежат генетически детерминированные нарушения сигнальных путей интерферонов I типа с повышением их продукции. Большинство дебютирует в детском возрасте. Клинические признаки включают лихорадку, кожные проявления, васкулопатии, интерстициальное поражение легких, поражение ЦНС, опорно-двигательного аппарата.
Интерферонопатия наследуется аутосомно-доминантным и аутосомно-рецессивным путем.
Большинство интерферонопатий наследуется аутосомно-рецессивным путем. В этом случаи родители, как правило, являются здоровыми носителями. Риск развития заболевания у ребенка составляет 25%, при аутосомно-доминантном типе наследования болен один из родителей, если это не мутация de novo, и в этом случае риск развития заболевания у ребенка составляет 50%.
В связи с чем рекомендовано проведении перинатальной диагностики при планировании следующей беременности.
Для диагностики интерферонопатий проводятся основные исследования: общий анализ крови с лейкоцитарной формулой, СОЭ, биохимия крови, определение уровня иммуноглобулинов, СРБ, определение аутоантител, интерферонового статуса в крови и ликворе, люмбальная пункция, с целью верификации диагноза проводится молекулярно-генетическое исследование. В последующем люмбальная пункция и определение уровня интерферонов проводятся по необходимости, исходя из тяжести заболевания.
Все пациенты с интерферонопатией нуждаются в терапии ингибитором JAK киназ. Препарат имеет таблетированную форму и принимается ежедневно от 4 до 5 раз в день. Доза зависит от тяжести течения заболевания и весо-ростовых показателей пациента. В случае неэффективности или недостаточном эффекте от проводимой терапии проводится коррекция дозы, а в последующем возможен альтернативный вариант в виде терапии моноклональными антителами к интерферону альфа.
Приложение Г
Нет.